
September 29, 2025
Daily life is all we need to sense that many factors influence our feelings, but the job of neuroscientists is to go beyond intuition with rigorous, experimental observation. That’s how they advance understanding. Via novel investigations of a variety of brain mechanisms that influence mood, several Picower Institute labs are advancing new ideas with the potential to address PTSD, anxiety, depression and bipolar disorder.
They are aware of the stakes. In the U.S., nearly 10 percent of adults experience a mood disorder in any given year, according to the National Institute of Mental Health. The need for better therapeutics and diagnostics is widespread and urgent.
For instance, in research stretching from 2014 to this year, Picower Professor Susumu Tonegawa has pinpointed exact brain circuits that associate the memory of an experience with a feeling about it. When his lab demonstrated in mice how that mechanism could be manipulated to change a negative feeling about a memory into a positive one, messages from people with anxiety disorders came flooding in, recalls longtime lab research scientist Michele Pignatelli. Never mind that the manipulation required genetically engineering the mice so that their neurons could be controlled by light from a fiber optic cable. People expressed eagerness to have fiber optics implanted in their brain.
The poignant pleas illustrated how pervasive and terrible disorders of mood can be. Tonegawa said he hopes his fundamental insights will find clinical application, and with the same motivation, three other Picower labs—those of Gloria Choi, Emery N. Brown and Elly Nedivi—are also studying novel mechanisms of mood.
Antagonism in the amygdala
Tonegawa’s research, pioneered in the lab by former graduate student Joshua Kim, centered on the amygdala brain region. Though the amygdala had already been established as a locus for emotion, his lab’s studies were the first to reveal both precisely and causally, at the molecular, cellular and circuit level, how the brain endows memories with emotional associations.

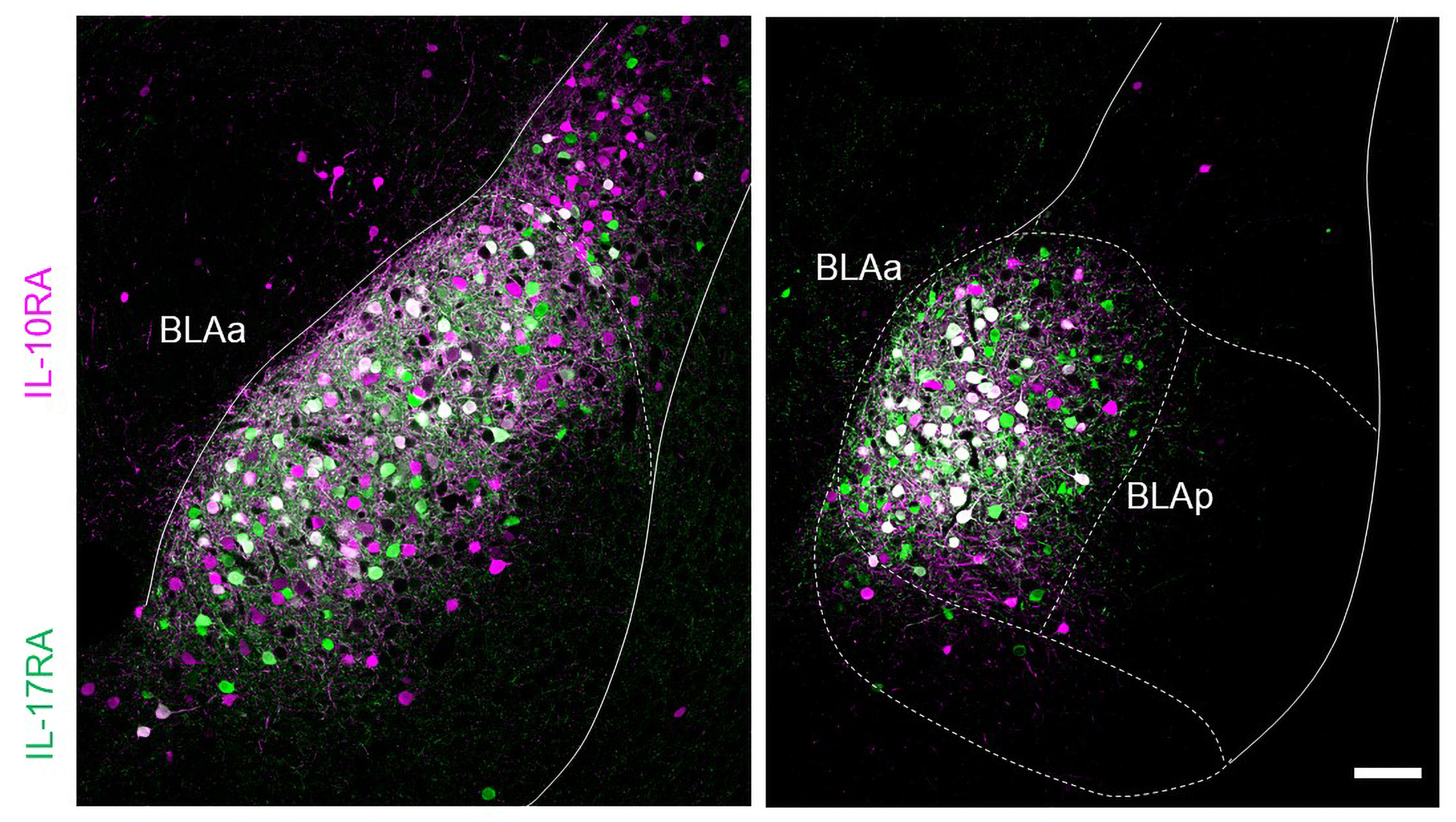
Kim and fellow lab member Roger Redondo set the table for the lab’s line of research with a 2014 study that took advantage of their recently perfected method for identifying the exact ensemble, or “engram,” of cells that encode a specific memory. Upon pinpointing engram cells, the scientists could then genetically engineer them with an emerging technology called optogenetics to make them controllable with light. Tonegawa’s team thereby gained the ability to take control of memories. In the study, Redondo and Kim labeled engrams in the brain’s hippocampus, which encodes memories of places, and the “basolateral” area of the amygdala (BLA) while exposing male mice to something like a modern-day twist on how Pavlov associated a conditioned stimulus (the sound of a bell) with an unconditioned one (a food reward). In Tonegawa’s case, the context was a place and the unconditioned stimulus was either a harmless but annoying shock or the company of a female mouse. The mice naturally associated the place memory with a good or bad feeling depending on the unconditioned stimulus and behaved accordingly when the researchers used their optogenetic control to reactivate the place memory. But then, Redondo, Kim and Tonegawa’s team reactivated the engram in the hippocampus while exposing the mice to the opposite unconditioned stimulus. When they did so, they successfully flipped the feelings the mice displayed for the place memory. And in the brains of the mice, the researchers could see that the place memory was now connecting with different cells in the amygdala than before.
The research team realized that the BLA not only endowed memories with their valence of good or bad feeling, but also that potentially different populations of cells in the region were responsible. To investigate that hypothesis, Kim led a new study in 2016. With Pignatelli and other members of Tonegawa’s lab, Kim employed clever molecular techniques to discover that the BLA contained two spatially separated populations of cells with distinct markers of gene expression and opposite functions. In the region’s posterior, cells that uniquely expressed the protein Ppp1r1b encoded positive valence, and cells in the anterior that uniquely expressed Rspo encoded negative valence. The study showed that these populations drove behavior. For instance, mice would show attraction to a place when posterior cells were optogenetically activated and aversion to a place when anterior cells were activated. Moreover, Kim discovered that the two BLA populations were locked in a circuit of mutual inhibition. Engaging one group suppressed the other. For example, mice exposed to a shock wouldn’t show as much fear if their posterior BLA cells were optogenetically activated because those cells would compete with the activity of the anterior cells that encoded fear of the shock.
That antagonism played out in a 2020 study in which Kim and graduate student Xiangyu Zhang tracked how the different populations battled during “fear extinction,” in which mice originally shocked in a place were later returned with no further shocks, ending the fear. Rspo2 cells were more active when the fear was learned and Ppp1r1b cells were more active during the fear extinction. Moreover, optogenetically activating Ppp1r1b cells accelerated fear extinction and suppressing the neurons made fear extinction harder, demonstrating that Ppp1r1b cell activity suppresses the valence encoded by Rspo2 cells.
“This phenomenon of fear extinction, it turns out, is not just the subsiding of an acquired fear memory,” Tonegawa said. “There is an active process, an active mechanism, to counter and abolish fears. Under some conditions, this fear extinction becomes very ineffective and that’s where we see a connection to post-traumatic stress disorder.”
The study also showed that the Ppp1r1b cells are the same neurons that encode the sense of reward, explaining why it feels so joyous when an expected fear doesn’t materialize (Tonegawa cites the elation students feel when a dreaded exam is canceled).
This past April, the lab answered another key question about this mechanism of mood and memory: What triggers it? Dopamine is known for signaling reward, and in the new study Zhang, former graduate student Kaetlyn Flick and Pignatelli rigorously proved that dopamine from the brain’s ventral tegmental area specifically cued each BLA population to assign negative valence to fear memories and positive valence to fear extinction memories.
In all, the lab has not only shown which BLA cells assign valence, but also that specific dopamine delivery circuits instigate that action, providing a molecular roadmap toward treatments for PTSD or anxiety.
Immune influence
Also in April, Associate Professor Gloria Choi’s lab published a study identifying an entirely different mechanism of mood—the immune system—that acts on the same Rspo-expressing population that Tonegawa characterized.
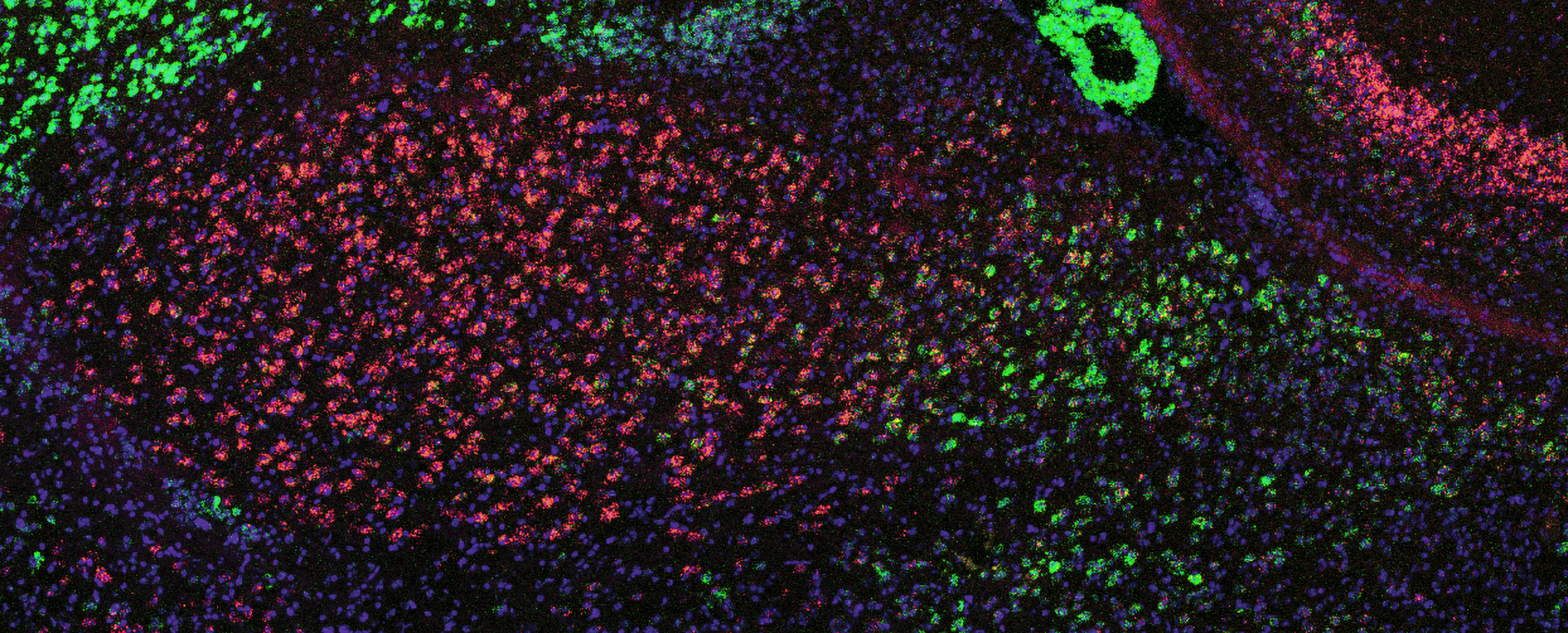
Before Choi began the study with collaborators at Harvard, doctors had observed that some people taking a psoriasis medication that suppresses the immune system had experienced severely negative thoughts. The medicine worked by blocking the “IL-17RA” receptor for inflammatory signaling molecules including IL-17C. Choi’s study, in mice, explained how that resulted in psychiatric symptoms. Blocking the receptor in the body left an excess of IL-17C to find its way to the brain. When IL-17C reaches the BLA, the study showed, receptors on Rspo neurons soaked in the molecule, activating the cells and producing anxious behaviors in the mice.
The study, co-led by postdoc Byeongjun Lee and former postdoc Jeong-Tae Kwon, also found that the cells have receptors for an anti-inflammatory molecule, IL-10. When they blocked a receptor of IL-10 to the anterior BLA neurons, the mice acted more anxiously, suggesting that IL-10 normally helps to keep anxiety in check.
Choi marvels at how the molecules’ opposing effects on the immune system (triggering inflammation or calming it) are mirrored in the amygdala.
“To have that system exactly reflected in the brain and to have the same population expressing these two different kinds of receptors so that when they are engaged they have opposite effects, I think, is so amazing and so beautiful,” Choi said. “It really is canonical evidence that these two systems, the brain and the immune, have evolved together to talk to each other.”
Why should they? We often withdraw and hunker down when we don’t feel well. That’s probably not just because of fatigue, Choi said. A little anxiety when we are unwell might protect us, perhaps by encouraging us to conserve energy and avoid danger in our weakened state. It also protects the community by reducing our desire to go near others. In fact, when we are sick, Choi said, the blood-brain barrier that normally filters what gets to the brain opens up potentially to facilitate this messaging from the peripheral immune system to the central nervous system.
Choi’s study raises key clinical questions. Should drugmakers be more wary that manipulating the immune system in the body might inadvertently manipulate the brain? And can the immune system be intentionally manipulated to treat mental health disorders? The key to both questions might be developing better ways to ensure that therapies only act where they are supposed to, Choi said.
“How do you target the brain but not the body and how can you target the body but not the brain?” Choi said. “That really is the homework for the pharmaceutical industry.”
Disrupting dynamics
Choi and Tonegawa acknowledge that there is more to the mechanisms they are studying, and to mood disorders in general. The brain is a complex network of networks linked both by direct neural connections and the flow of neuromodulatory chemicals.
For instance, in a review paper published in February, Edward Hood Taplin Professor Emery N. Brown and co-authors noted three broad-based theories of depression as they explained why eight different anesthetic drugs might be effective antidepressants (so far only ketamine is used that way).
One theory holds that depression arises from perturbations of brain networks that normally collaborate to regulate emotion and attention to internal thought vs. external stimuli. Another theory focuses on a deficit of BDNF, a brain chemical that promotes increased connectivity among neurons. Yet another theory focuses on the inhibitory neurotransmitter GABA, suggesting that reduced levels can promote depression when neurons become overexcited.
After noting that many different anesthetic drugs have been experimentally (and anecdotally) observed to relieve depression, Brown’s review describes how drugs including ketamine, propofol, and dexmedetomidine directly intersect with the mechanisms underlying these theories. Ketamine increases BDNF and alters network connectivity. Propofol works by increasing GABA and it both increases BDNF and alters broader network connectivity. Dexmedetomidine also increases BDNF and reduces broad network connectivity.
“The common factor among all the anesthetics is that they change brain dynamics,” the review states. “We propose that the therapeutic actions of anesthetics in treating major depressive disorder or treatment-resistant depression could be their capacity to disrupt and help rewire brain networks.”
Many details remain to be discovered. Brown takes inspiration from an old but nonetheless effective way of disrupting intractably depressed brain networks: electroconvulsive, or “shock,” therapy (ECT). On the way of understanding whether and how different anesthetics might do a better job and cause fewer cognitive side effects, Brown’s lab led by postdoctoral Picower Fellow Macauley Breault and senior postdoctoral associate Sirma Orguc are now gathering data from ECT patients at McLean Hospital near Boston. They’ll seek to determine a more detailed understanding of how ECT and the different anesthetics given to patients during the procedure combine to alter brain networks.
“What we’d like to do is at least have some idea of what the comparator (ECT) is doing,” Brown said. “If we do a clinical trial where one group gets anesthesia alone and the other group gets ECT, then at least we’ll have some idea of what we should be looking for.”
A lead on bipolar disorder
William and Linda Young Professor Elly Nedivi is on the trail of another mechanism of mood: genetic risk for bipolar disorder.
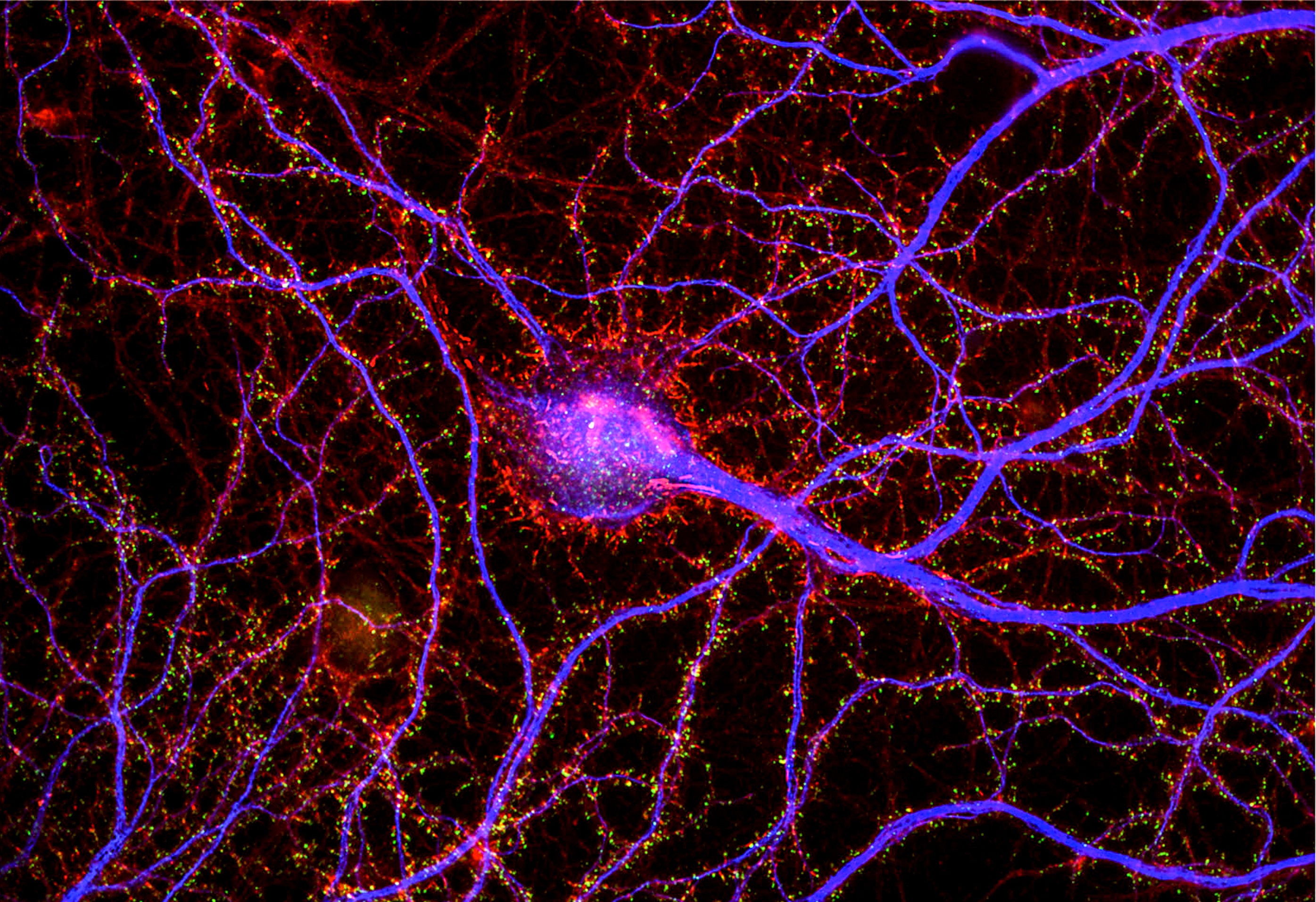
In 2019, Nedivi’s lab, led by former postdoc Mette Rathje, published a study showing that people diagnosed with bipolar disorder had lower levels in the brain of the CPG2 protein, a product of the SYNE1 gene which had been established as a genetic hit for bipolar disorder. They then identified specific patient mutations (or combinations of them) in the part of SYNE1 controlling CPG2 expression and showed that they led to reduced expression of CPG2.
The study raised the possibility that testing people for particular mutations of SYNE1 could be a biomarker of bipolar disorder risk. Such an objective marker could help increase confidence when diagnosing bipolar, Nedivi said. Misdiagnosis can lead to patients receiving inappropriate treatment, sometimes for years.
“The diagnostic is entirely subjective through symptoms,” Nedivi said. “But there is a lot of overlap between bipolar symptoms and either schizophrenia or major depression. So when patients come to a doctor, depending on what symptoms they have at the time, the diagnosis is not always simple. Having an objective biological diagnostic would make the whole treatment journey potentially a little less fraught.
Nedivi originally discovered CPG2 decades ago as part of fundamental studies of how neurons forge and edit their circuit connections, or “synapses,” to enable the brain to flexibly respond to experience, a phenomenon known as “plasticity.” CPG2 does this by regulating the number of receptors for the neurotransmitter glutamate at excitatory synapses. This doesn’t immediately explain why a deficit could increase bipolar risk, but it does provide some clues. Like BNDF, CPG2 affects neural connectivity and its expression depends on neural activity, Nedivi said. CPG2’s effects on the refinement of neural circuits also kicks in very late in brain development (i.e. adolescence), when bipolar onset often occurs. And finally, CPG2 expression is more prominent in areas of the brain related to cognition.
Since the 2019 study, the lab led by postdoctoral Picower Fellow Baovi Vo has been working to further bolster the evidence base for which particular SYNE1 mutations most contribute to CPG2 expression anomalies. To do that, they are digging deep into a database of human cell cultures derived from thousands of people. That effort will vastly increase the lab’s sample size, increasing the reliability of potential genetic biomarkers.
From sources as diverse as memory, immune activity, anesthesia and synaptic plasticity genes, many mechanisms converge on mood. Those Picower Institute insights can inform new ways to improve mental health.



