
September 24, 2020
Their words reached what may be the biggest audience ever for a Picower Institute symposium. More than 1,300 people from 48 countries tuned in online over the course of the day.
“Neurodegeneration is a very multifaceted and difficult problem that affects tens of millions of people around the world,” said Li-Huei Tsai, Picower Professor of Neuroscience, director of The Picower Institute for Learning and Memory, and founding director of the ABI. “The good news is that many very talented and dedicated researchers are working on it.”
Indeed, speaker after speaker throughout the day not only shared new insights into the nature of diseases that cause brain cells to die, but also described promising new treatment strategies. Many are advancing through clinical trials.
Keynote speaker Don Cleveland of the University of California at San Diego, for example, won the Breakthrough Prize in 2018 for advancing a technology called antisense oligonucleotide therapy, which involves synthesizing “designer DNA drugs” that can intercede to compensate for a genetic mutation, either to reduce the production of harmful proteins, or to enhance production of needed proteins. He described how his lab helped to pioneer the idea for ALS in 2006 and how, working with Ionis Pharmaceuticals, it’s being applied to treat spinal muscular atrophy and is now being tested for Huntington’s disease and ALS. He added that in some of his lab’s most recent work, he is testing a potential treatment for Parkinson’s disease in which brain cells called astrocytes can be converted into neurons in a key brain region by lowering their expression of a gene called PTB. Data in mice show that after this “identity theft,” as Cleveland termed it, the newly converted neurons could take the place of neurons that died during disease, restoring the essential function of producing the neurotransmitter dopamine.
Above: Lennart Mucke (bottom center) speaks as part of a panel discussion with fellow speakers Anne Brunet, Li-Huei Tsai, Don Cleveland and Valina Dawson.
Each researcher who spoke is tackling a different aspect of the broad spectrum of scientific questions that surround neurodegeneration— focusing on dysfunctions at the level of genes, proteins, cells or systems.
Diane Chan, a neurologist and postdoctoral researcher in Tsai’s Picower Institute lab, is leading human clinical studies of a potential Alzheimer’s therapy that addresses brain dysfunction at the level of whole-brain network activity. After observing that the power of neural rhythms at “gamma” frequency of 40 Hz are lessened in Alzheimer’s, Tsai’s lab led the discovery that exposing mice to light or sound at that frequency can increase gamma power and restore synchrony of neural activity across brain regions. In mouse models the technique improves memory and cognition, prevents neural death and reduces the accumulation of proteins known as key biomarkers of disease progression. The exact mechanisms are not fully understood, Chan said, but the stimulation appears to restore healthier gene expression and activity to neurons and brain immune cells called microglia, and causes brain blood vessels to widen.
In a small sample of human volunteers tested so far, the method has proven safe and well tolerated, increases 40 Hz gamma rhythm power and network connectivity and may be contributing to improved sleep, Chan reported. Testing for improvements in memory and cognition have not been completed yet, however.
Microglia cells also proved central in talks by both Dorothy Schafer of the University of Massachusetts Medical School and Marco Colonna of Washington University. In a mouse model of multiple sclerosis called EAE, Schafer has pinpointed the molecular process by which microglia end up engulfing and destroying the connections between neurons, called synapses. Based on those findings, her team has shown that overexpression of a protein that specifically blocks that action preserves synapses and protects brain functions, such as vision, that deteriorate in untreated mice modeling EAE.
While Schafer reported progress in understanding how to reduce a harmful activity of microglia, Marco Colonna of Washington University discussed his work to overcome genetic variants that appear to prevent microglia from doing their job of removing potentially harmful proteins. He’s shown that in people with particular variants of the gene Trem2, microglia fail to surround and engulf amyloid beta protein as they should. Their lack of action, he said, may not cause Alzheimer’s but accelerates the resulting disease progression. In his lab’s work with animals he’s found an antibody that stimulates microglia with these deficiencies to spring back into action, reducing amyloid plaques and preventing damage to neurons.
Colonna’s Washington University colleague Jonathan Kipnis illustrated a very different immune system role in neurodegenerative disease. He highlighted his lab’s recent findings that immune cells patrol around the brain in the meninges, the space outside the brain but within the skull. There, they’ve been studying vessels that connect the brain to the lymphatic system, allowing for circulation and drainage of waste-carrying cerebrospinal fluid from the brain. His lab has recently found that if meningeal lymphatic vessels are impaired, as they can be in older mice, then antibodies meant to remove amyloid plaques from the brain provide no benefit. But they have also shown that treating lymphatic vessels in the brain’s meninges with the growth factor VEGF-C can improve the effect of anti-amyloid antibodies, even in older mice.
Much as the lymphatic system allows for clearance of waste the circulatory system brings in needed nutrients. In his talk David Attwell of University College London showed that early in Alzheimer’s disease upon exposure to amyloid protein, cells called pericytes begin to constrict blood flow through capillaries of the brain, depriving cells of needed energy. In Alzheimer’s model mice, his lab has looked for the molecular culprits that mediate this phenomenon and found ways to block the pathway they follow, restoring fuller capillary diameter.
The blood and lymphatic systems are not the only ways the brain has exposure to the body. The peripheral nervous system also provides a path. Valina Dawson of Johns Hopkins University showed how toxic forms of a protein called alpha synuclein might invade the brain by traveling from the gut through the vagus nerve, causing alpha synuclein already there to misfold, explaining how it can lead to Parkinson’s disease damage, even in animals (or people) who don’t have any known genetic risk. Dawson’s lab has also identified the receptor that lets toxic alpha synuclein into neurons and has shown that blocking that receptor protects cells.
Misfolded or abnormally aggregated proteins are often implicated in neurodegenerative disease.
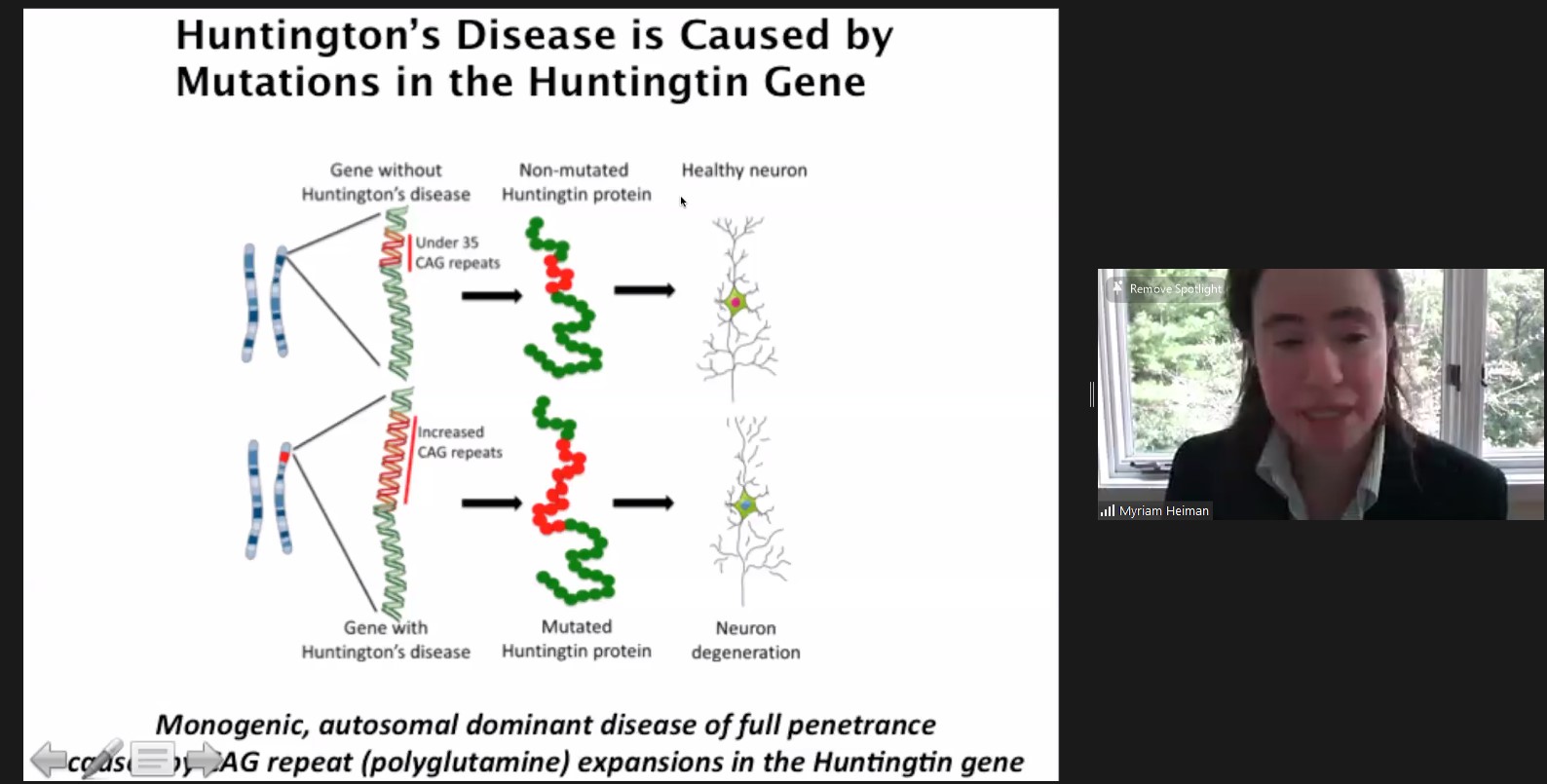
In her studies of Huntington’s, Picower Institute member Myriam Heiman, Associate Professor of Brain and Cognitive Sciences at MIT, has sought to understand why specific cells in a region of the brain called the striatum are especially susceptible to an aggregation of huntingtin. To find out, she engaged in an unbiased genetic screen of the entire mouse genome, looking for genes that, if knocked out, would make neurons carrying the Huntington’s mutation especially vulnerable. In her talk she described the findings of her study, published in January in Neuron that found several genes including a novel one, Nme1. Further investigation revealed that Nme1 likely helps promote clearance of huntingtin aggregates. In disease model mice, she showed that overexpressing Nme1 reduced disease symptoms.
Stanford University’s Anne Brunet discussed two lines of research regarding normal aging. In one, she found that T cells accumulate in the brains of older mice, negatively impacting the proliferation of neural stem cells and therefore their ability to produce new neurons and repair the brain. In the other, newer line of work, she is finding that in African killifish, which model vertebrate aging, there may be an age-dependent propensity for aggregation of many proteins in different organs, including the brain. The aggregation made even worse if they had an advanced aging disease she modeled by mutating their telomerases. The accumulation of a particular protein that she found in the brains of both the fish and in mice, may cause further protein aggregation that affects brain function.
One of the proteins whose aberrant behavior is most commonly associated with neurodegeneration is tau. Discovered by Cleveland in the 1970s, tau is now studied in many labs. At the symposium, leading tau researcher Lennart Mucke of the University of California at San Francisco presented evidence that in both Alzheimer’s and even some autism spectrum disorders, tau can still enable dysfunction in disease processes, even when tau itself is in a healthy form. For instance, he’s found that the protein is necessary for neuronal hyperexcitability to emerge in Alzheimer’s disease models and in the autism disorder Dravet syndrome. Reducing the protein helps prevent epileptic activity in many cases, he said, and reduces other symptoms in some but not other autism mouse models. The research suggests that lowering overall healthy tau levels might have therapeutic benefits in some diseases.
Twice during the day the speakers gathered in panels to engage each other in direct discussion. In the latter of those, Mucke offered a summary of the day that struck a hopeful note about the diversity of new progress, but also called upon pharmaceutical companies to resume more neurodegeneration research, so they can help develop the most clinically relevant ideas coming out of academic research.
“Looking at the whole day, I am impressed with the many, many good leads that exist in neurodegeneration that could possibly contribute to what matters the most to patients, which is the dysfunction of their brain,” he said. “One can make so many great cases for so many things, but the key question is: What is their relative contribution that they are making to two things—brain dysfunction that clinically matters and what difference does it make to the demise of neurons?”



