
March 14, 2022
Muscles and bones strengthen with exercise and the immune system ‘learns’ from vaccines or infections, but none of those changes match the versatility and flexibility your central nervous system shows in adapting to the world. The brain is a model remodeler. If it weren’t, you wouldn’t have learned how to read this and you wouldn’t remember it anyway.
The brain’s ability to change its cells, their circuit connections, and even its broader architectures in response to experience and activity, for instance to learn new rules and store memories, is called “plasticity.” The phenomenon explains how the brand-new brain of an infant can emerge from a womb and make increasingly refined sense of whatever arbitrary world it encounters – ranging from tuning its visual perception in the early months to getting an A in eighth-grade French. Plasticity becomes subtler during adulthood, but it never stops. It occurs via so many different mechanisms and at so many different scales and rates, it’s… mind-bending.
Plasticity’s indispensable role in allowing the brain to incorporate experience has made understanding exactly how it works – and what the mental health ramifications are when it doesn’t – the inspiration and research focus of several Picower Institute professors (and hundreds of colleagues). This site uses the term so often in reports on both fundamental neuroscience and on disorders such as autism, it seemed high time to provide a primer. So here goes.
Beginning in the 1980s and 1990s, advances in neuroanatomy, genetics, molecular biology and imaging made it possible to not only observe, but even experimentally manipulate mechanisms of how the brain changes at scales including the individual connections between neurons, called synapses; across groups of synapses on each neuron; and in whole neural circuits. The potential to discover tangible physical mechanisms of these changes proved irresistible to Picower Institute scientists such as Mark Bear, Troy Littleton, Elly Nedivi and Mriganka Sur.
Bear got hooked by experiments in which by temporarily covering one eye of a young animal, scientists could weaken the eye’s connections to the brain just as their visual circuitry was still developing. Such “monocular deprivation” produced profound changes in brain anatomy and neuronal electrical activity as neurons rewired circuits to support the unobstructed eye rather than the one with weakened activity.
“There was this enormous effect of experience on the physiology of the brain and a very clear anatomical basis for that,” Bear said. “It was pretty exhilarating.”
Littleton became inspired during graduate and medical school by new ways to identify genes whose protein products formed the components of synapses. To understand how synapses work was to understand how neurons communicate and therefore how the brain functions.
“Once we were able to think about the proteins that are required to make the whole engine work, we could figure out how you might rev it up and down to encode changes in the way the system might be working to increase or decrease information flow as a function of behavioral change,” Littleton said.
Built to rebuild
So what is the lay of the land for plasticity? Start with a neuron. Though there are thousands of types, a typical neuron will extend a vine-like axon to forge synapses on the root-like dendrites of other neurons. These dendrites may host thousands of synapses. Whenever neurons connect, they form circuits that can relay information across the brain via electrical and chemical signals. Most synapses are meant to increase the electrical excitement of the receiving neuron so that it will eventually pass a signal along, but other synapses modulate that process by inhibiting activity.
Hundreds of proteins are involved in building and operating every synapse, both on the “pre-synaptic” (axonal) side and the “post-synaptic” (dendritic) side of the connection. Some of these proteins contribute to the synapse’s structure. Some on the pre-synaptic side coordinate the release of chemicals called neurotransmitters from blobs called vesicles, while some on the postsynaptic side form or manage the receptors that receive those messages. Neurotransmitters may compel the receiving neuron to take in more ions (hence building up electric charge), but synapses aren’t just passive relay stations of current. They adjust in innumerable ways according to changing conditions, such as the amount of communication activity the host cells are experiencing. Across many synapses the pace and amount of neurotransmitter signaling can be frequently changed by either the presynaptic or postsynaptic side. And sometimes, especially early in life, synapses will appear or disappear altogether.
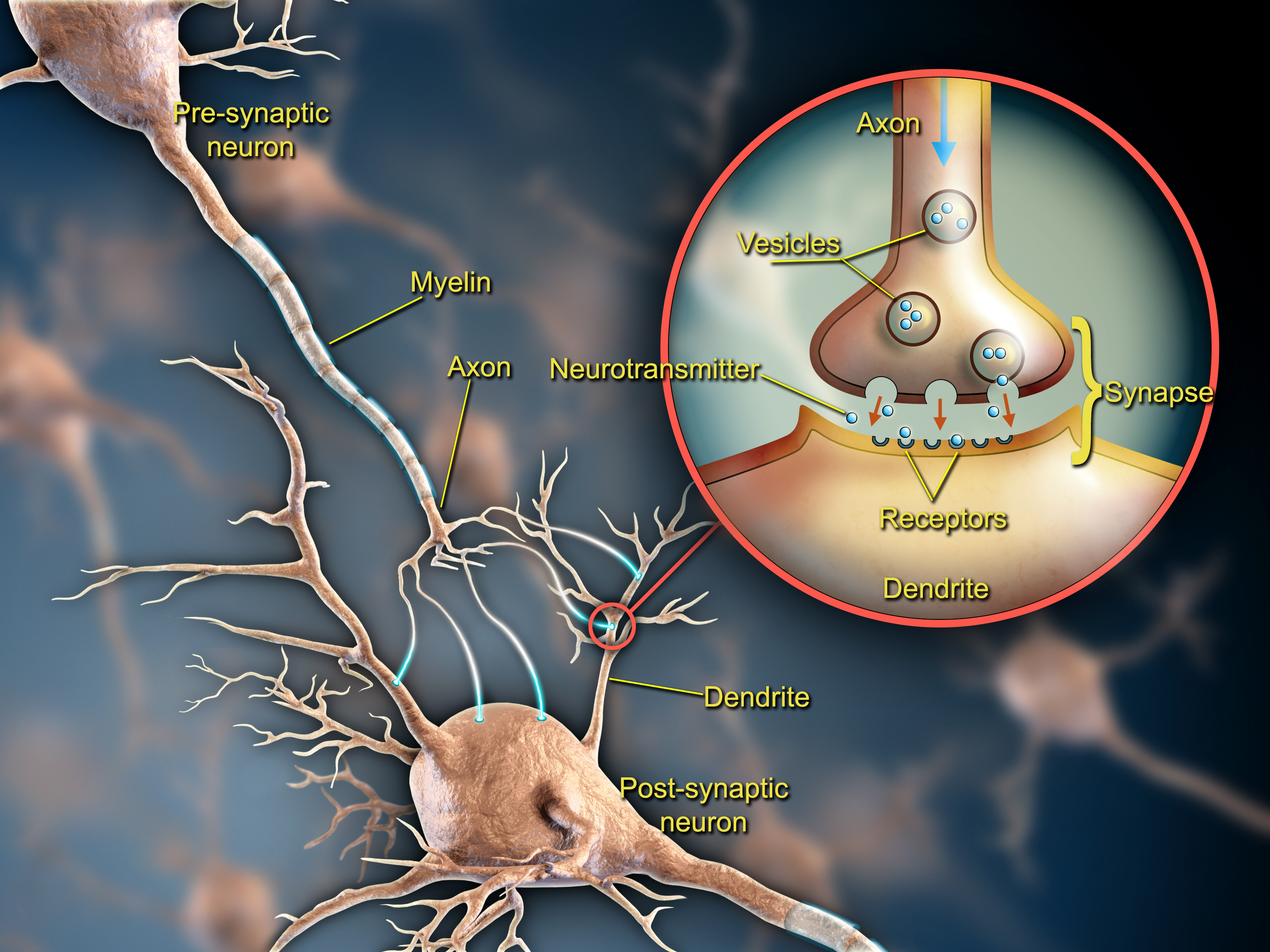
Moreover, plasticity doesn’t just occur at the level of the single synapse. Combinations of synapses along a section of dendrite can all change in coordination so that the way a neuron works within a circuit is altered. These numerous dimensions of plasticity help to explain how the brain can quickly and efficiently accomplish the physical implementation of something as complex as learning and memory, Nedivi said.
“You might think that when you learn something new it has nothing to do with individual synapses,” Nedivi said. “But in fact, the way that things like this happen is that individual synapses can change in strength or can be added and removed, and then it also matters which synapses, and how many synapses, and how they are organized on the dendrites, and how those changes are integrated and summated on the cell. These parameters will alter the cell’s response properties within its circuit and that affects how the circuit works and how it affects behavior.”

A 2018 study in Sur’s lab illustrated learning occurring at a neural circuit level. His lab trained mice on a task where they had to take a physical action based on a visual cue (e.g. drivers know that “green means go”). As mice played the game, the scientists monitored neural circuits in a region called the posterior parietal cortex where the brain converts vision into action. There, ensembles of neurons increased activity specifically in response the “go” cue. When the researchers then changed the game’s rules (i.e. “red means go”) the circuits switched to only respond to the new go cue. Plasticity had occurred en masse to implement learning.
Many mechanisms
To carry out that rewiring, synapses can change in many ways. Littleton’s studies of synaptic protein components have revealed many examples of how they make plasticity happen. Working in the instructive model of the fruit fly, his lab is constantly making new findings that illustrate how changes in protein composition can modulate synaptic strength.
For instance, in a 2020 study his lab showed that synaptotagmin 7 limits neurotransmitter release by regulating the speed with which the supply of neurotransmitter-carrying vesicles becomes replenished. By manipulating expression of the protein’s gene, his lab was able to crank neurotransmitter release, and therefore synaptic strength, up or down like a radio volume dial.
Other recent studies revealed how proteins influence the diversity of neural plasticity. At the synapses flies use to control muscles, “phasic” neurons release quick, big bursts of the neurotransmitter glutamate, while tonic ones steadily release a low amount. In 2020 Littleton’s lab showed that when phasic neurons are disrupted, tonic neurons will plasticly step up glutamate release, but phasic ones don’t return the favor when tonic ones are hindered. Then last year, his team showed that a major difference between the two neurons was their levels of a protein called tomosyn, which turns out to restrict glutamate release. Tonic ones have a lot but phasic ones have very little. Tonic neurons therefore can vary their glutamate release by reducing tomosyn expression, while phasic neurons lack that flexibility.
Nedivi, too, looks at how neurons use their genes and the proteins they encode to implement plasticity. She tracks “structural plasticity” in the living mouse brain, where synapses don’t just strengthen or weaken, but come and go completely. She’s found that even in adult animal brains, inhibitory synapses will transiently appear or disappear to regulate the influence of more permanent excitatory synapses.
Nedivi has revealed how experience can make excitatory synapses permanent. After discovering that mice lacking a synaptic protein called CPG15 were slow learners, Nedivi hypothesized that it was because the protein helped cement circuit connections that implement learning. To test that, her lab exposed normal mice and others lacking CPG15 to stretches of time in the light, when they could gain visual experience, and the dark, where there was no visual experience. Using special microscopes to literally watch fledgling synapses come and go in response, they could compare protein levels in those synapses in normal mice and the ones without CPG15. They found that CPG15 helped experience make synapses stick around because upon exposure to increased activity, CPG15 recruited a structural protein called PSD95 to solidify the synapses. That explained why CPG15-lacking mice don’t learn as well: they lack that mechanism for experience and activity to stabilize their circuit connections.
Another Sur Lab study in 2018 helped to show how multiple synapses sometimes change in concert to implement plasticity. Focusing on a visual cortex neuron whose job was to respond to locations within a mouse’s field of view, his team purposely changed which location it preferred by manipulating “spike-timing dependent plasticity.” Essentially right after they put a visual stimulus in a new location (rather than the neuron’s preferred one), they artificially excited the neuron. The reinforcement of this specifically timed excitement strengthened the synapse that received input about the new location. After about 100 repetitions, the neuron changed its preference to the new location. Not only did the corresponding synapse strengthen, but also the researchers saw a compensatory weakening among neighboring synapses (orchestrated by a protein called Arc). In this way, the neuron learned a new role and shifted the strength of several synapses along a dendrite to ensure that new focus.
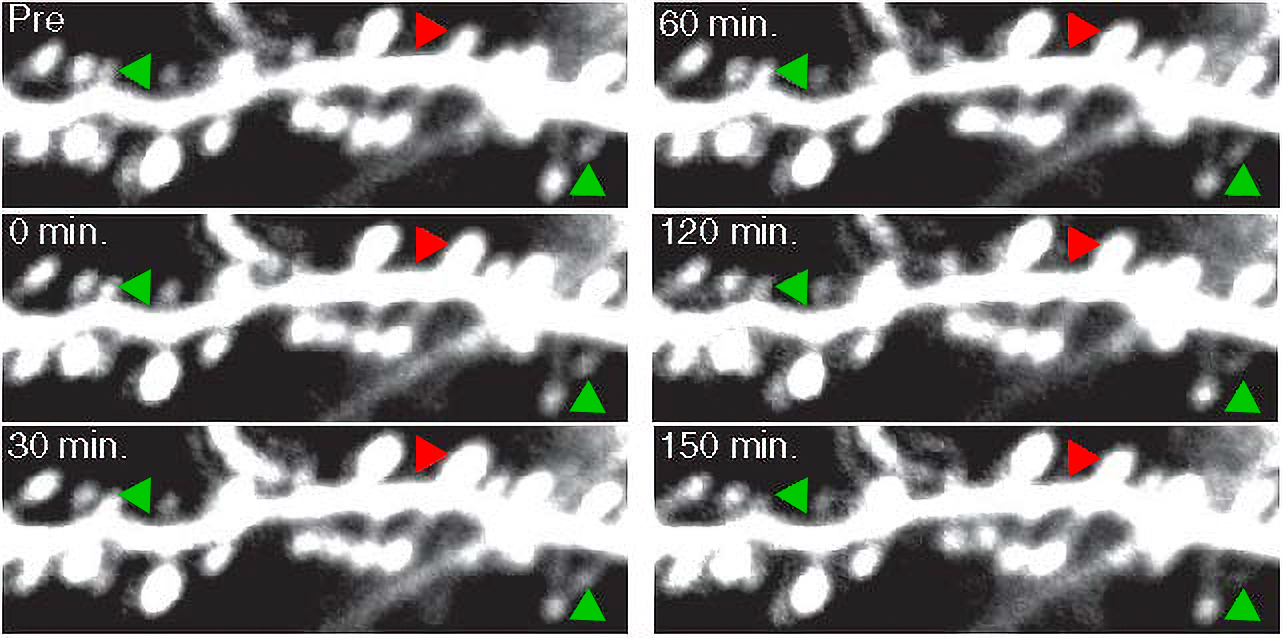
Lest one think that plasticity is all about synapses or even dendrites, Nedivi has helped to show that it isn’t. For instance, her research has shown that amid monocular deprivation, inhibitory neurons go so far as to pare down their axons to enable circuit rewiring to occur. In 2020 her lab collaborated with Harvard scientists to show that to respond to changes in visual experience, some neurons will even adjust how well they insulate their axons with a fatty sheathing called myelin that promotes electrical conductance. The study added strong evidence that myelination also contributes to the brain’s adaptation to changing experience.
It’s not clear why the brain has evolved so many different ways to effect change (these examples are but a small sampling) but Nedivi points out a couple of advantages: robustness and versatility.
“Whenever you see what seems to you like redundancy it usually means it’s a really important process. You can’t afford to have just one way of doing it,” she said. “Also having multiple ways of doing things gives you more precision and flexibility and the ability to work over multiple time scales, too.”
Insights into illness
Another way to appreciate the importance of plasticity is to recognize its central role in neurodevelopmental diseases and conditions. Through their fundamental research into plasticity mechanisms, Bear, Littleton, Nedivi and Sur have all discovered how pivotal they are to breakdowns in brain health.
Beginning in the early 1990s, Bear led pioneering experiments showing that by multiple means, post-synaptic sensitivity could decline when receptors received only weak input, a plasticity called long-term depression (LTD). LTD explained how monocular deprivation weakens an occluded eye’s connections to the brain. Unfortunately, this occurs naturally in millions of children with visual impairment, resulting in a developmental vision disorder called amblyopia. But Bear’s research on plasticity, including mechanisms of LTD, has also revealed that plasticity itself is plastic (he calls that “metaplasticity”). That insight has allowed his lab to develop a potential new treatment in which by completely but temporarily suspending all input to the affected eye by anesthetizing the retina, the threshold for strengthening vs. weakening can be lowered such that when input resumes, it triggers a newly restorative connection.
Bear’s investigations of a specific form of LTD have also led to key discoveries about Fragile X syndrome, a genetic cause of autism and intellectual disability. He found that LTD can occur when stimulation of metabotropic glutamate receptor 5 (mGluR5) causes proteins to be synthesized at the dendrite, reducing post-synaptic sensitivity. A protein called FMRP is supposed to be a brake on this synthesis but mutation of the FMR1 gene in Fragile X causes loss of FMRP. That can exaggerate LTD in the hippocampus, a brain region crucial for memory and cognition. The insight has allowed Bear to advance drugs to clinical trials that inhibit MGlur5 activity to compensate for FMRP loss.
Littleton, too, has produced insight into autism by studying the consequences of mutation in the gene Shank3, which encodes a protein that helps to build developing synapses on the post-synaptic side. In a 2016 paper his team reported multiple problems in synapses when Shank was knocked out in fruit flies. Receptors for a key form of molecular signaling from the presynaptic side called Wnt failed to be internalized by the postsynaptic cell, meaning they could not influence the transcription of genes that promote maturation of the synapse as they normally would. A consequence of disrupted synaptic maturation is that a developing brain would struggle to complete the connections needed to efficiently encode experience and that may explain some of the cognitive and behavioral outcomes in Shank-associated autism. To set the stage for potential drug development, Littleton’s lab was able to demonstrate ways to bypass Wnt signaling that rescued synaptic development.
By studying plasticity proteins Sur’s lab, too, has discovered a potential way to help people with Rett syndrome, a severe autism-like disorder. The disease is caused by mutations in the gene MECP2. Sur’s lab showed that MECP2’s contribution to synaptic maturation comes via a protein called IGF1 that is reduced among people with Rett. That insight allowed them to show that treating Rett-model mice with extra IGF1 peptide or IGF1 corrected many defects of MECP2 mutation. Both treatment forms have advanced to clinical trials. Late last year IGF1 peptide was shown to be effective in a comprehensive phase 3 trial for Rett syndrome and is progressing toward FDA approval as the first-ever mechanism-based treatment for a neurodevelopmental disorder, Sur said.
Nedivi’s plasticity studies, meanwhile, have yielded new insights into bipolar disorder. During years of fundamental studies, Nedivi discovered CPG2, a protein expressed in response to neural activity that helps regulate the number of glutamate receptors at excitatory synapses. The gene encoding CPG2 was recently identified as a risk gene for bipolar disorder. In a 2019 study her lab found that people with bipolar disorder indeed had reduced levels of CPG2 because of variations in the SYNE1 gene. When they cloned these variants into rats, they found they reduced the ability of CPG2 to locate in the dendritic “spines” that house excitatory synapses or decreased the proper cycling of glutamate receptors within synapses.
The brain’s ever-changing nature makes it both wonderful and perhaps vulnerable. Both to understand it and heal it, neuroscientists will eagerly continue studying its plasticity for a long time to come.



