Mindful of both its power and its limits, Picower Institute scientists are using single cell genomics techniques to measure gene expression and produce unique insights into nervous system biology and diseases such as Alzheimer’s and Huntington’s
When Biology Assistant Professor Sara Prescott was a new postdoc at Harvard in 2016, her lab had made an important advance in studying the vagus nerve, a major conduit of signals between the brain and the body’s organs. The lab had identified six different types of sensory neurons in the nerve bundle, validating the hypothesis that a diversity of vagal neurons sensed problems in the body and responded with appropriate reflexes.
When Prescott picked up the torch, she did so with a new tool called “single cell RNA sequencing” (scRNA-seq). Almost all cells in an organism have the same DNA, but each type of cell becomes distinct in its form and function by how it expresses those genes. scRNA-seq tallies up which genes a cell has transcribed into RNA, for instance to make a protein, and how often. At the time, scRNA-seq commercial systems weren’t widely available. Using it took considerable work. But when Prescott was done, her team showed that there were not just six distinct types of sensory neurons in the vagal nerve, but 37. The results she published in Cell in April 2020 have already been cited more than 100 times. The results continue to propel her work at MIT as she investigates the function of those neurons.
Back then Picower Professor and Institute Director Li-Huei Tsai had also just begun to incorporate scRNA-seq into her Alzheimer’s disease research. In 2017, she co-led its first use to analyze the brain’s microglia immune cells in Alzheimer’s disease model mice. Inflammation is a hallmark of Alzheimer’s pathology and this study showed how microglia become more inflammatory as the disease progresses. Since then Tsai has become a leader of using scRNAseq and other single cell genomic measures to discover how different cell types and molecular pathways go awry in Alzheimer’s disease, which ones contribute to resilience, and the effects of carrying Alzheimer’s disease risk genes or experiencing age-related DNA damage. In some of her most recent studies, she and collaborators also employed single cell ATAC-seq, which measures, cell by cell, how accessible genes are for transcription, providing a deeper understanding of how cells are regulating gene expression.
‘A readout and a reference’
Increasingly Picower Institute scientists are embracing the power (and working through the limitations) of single cell genomics techniques to make many influential discoveries about the diversity of cells in the nervous system and the way they each function in health and disease. By tracking indicators of gene expression cell by cell, they allow scientists to not only discern the cell types in a tissue but also how they are uniquely functioning, or faltering.
“Having a reference atlas of what's in the brain is very important, not just for the sake of knowledge and cataloging, but also to understand changes that might be occurring in normal development as well as in disease,” said Associate Professor Myriam Heiman, who has used scRNA-seq in several studies since 2020 that have advanced understanding of which cells are especially vulnerable in several neurodegenerative diseases and what makes them so.
Prescott agrees: “It is both a description of the [cell-type-specific] data that you have but it’s also a very valuable resource for doing follow-up studies on the functions of those cells. It’s a readout and a reference.”
The technique’s popularity has surged as it has become much easier to obtain scRNA-seq data from a sample, said Menicon Professor Troy Littleton, whose lab published its first two RNA sequencing papers this year as the team sought to investigate why two similar-seeming types of neurons exhibit distinct communication properties. But it’s no panacea, he said. The method can oblige users to perform detailed computational analyses and experimental investigation at the lab bench to, for instance, avoid falling for irrelevant hits that seem statistically significant but turn out to make no difference to a study’s hypothesis. But given the relative ease of getting the data, he said, there’s no reason not to take advantage of it when it can provide useful leads.
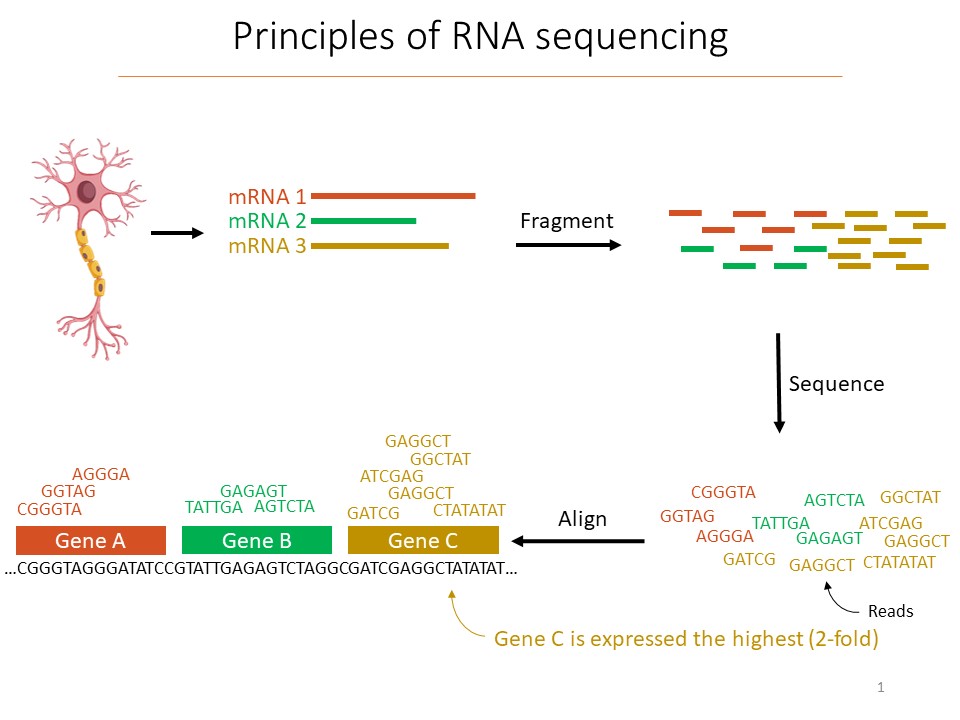
The promise and the pitfalls of scRNA-seq are explained by how it works. When scientists have a tissue sample, they first separate out each cell. The cells (or their nuclei) are then enveloped in a droplet and broken open. Pieces of the RNA that were being transcribed at the time are then “barcoded” with synthetic RNA that labels it uniquely and notes which cell it came from. The RNA fragments are then sequenced, which allows for identifying which gene they were transcribed from. In this way, for the organism’s whole genome, scientists get an unbiased accounting of which genes each cell in the sample was actually making use of, or “expressing,” and how much they were doing it.
In single cell RNA sequencing, the messenger RNA that a cell has transcribed from DNA is extracted and sequenced to form "reads." By matching reads to a reference genome, scientists can determine wich genes are being expressed most. Diagram courtesy of Andres Crane.
In a review paper in Nature Neuroscience in 2023, Tsai and graduate student Mitch Murdock noted that Alzheimer’s disease studies using postmortem human brains require careful thought about which samples to sequence. What stage of disease was the tissue obtained during? What are the demographics and lifestyle factors of the donors? Are the samples of good quality and are they processed right? The larger and more variable the samples are, the more tricky and difficult analysis will be to ensure that the results are biologically meaningful and not artifacts of flawed input. Tsai and Heiman, for instance, frequently work with MIT Computer Science Professor Manolis Kellis who is a globally renowned computational biology expert.
Even after expert analysis, one should still be skeptical of single cell genomics data alone. A significant difference in gene expression could arise for many reasons. Littleton found hundreds of differentially expressed genes between the two neurons his team was comparing, but in many cases the expression differences did not explain any of the differences between neurons he was interested in. Moreover, proteins his lab had already shown to be consequentially different between the two cells showed no difference in RNA levels. The differences arose from some other mechanism scRNA-seq doesn’t detect.
With the caveats in mind, Picower scientists have succeeded in using single cell genomics to catalog and understand the diversity of cells in the nervous system and to uncover mechanisms of how specific cellular functions become compromised in neurodegenerative diseases.
Cell diversity
Discovering and cataloging the diversity of cell types in a tissue is often called making a map or an atlas. With about 25,000 vagal sensory neurons from 40 mice, Prescott’s postdoctoral team was able to define their distinct expression profiles and discern which “markers” sorted them into 37 distinct clusters. Then they exploited those markers to distinctly label each kind of neuron and to experimentally manipulate them. Those steps allowed her to show that one of the neuron types she identified was critical for triggering life-saving swallowing and coughing reflexes when water or acid entered the throat.
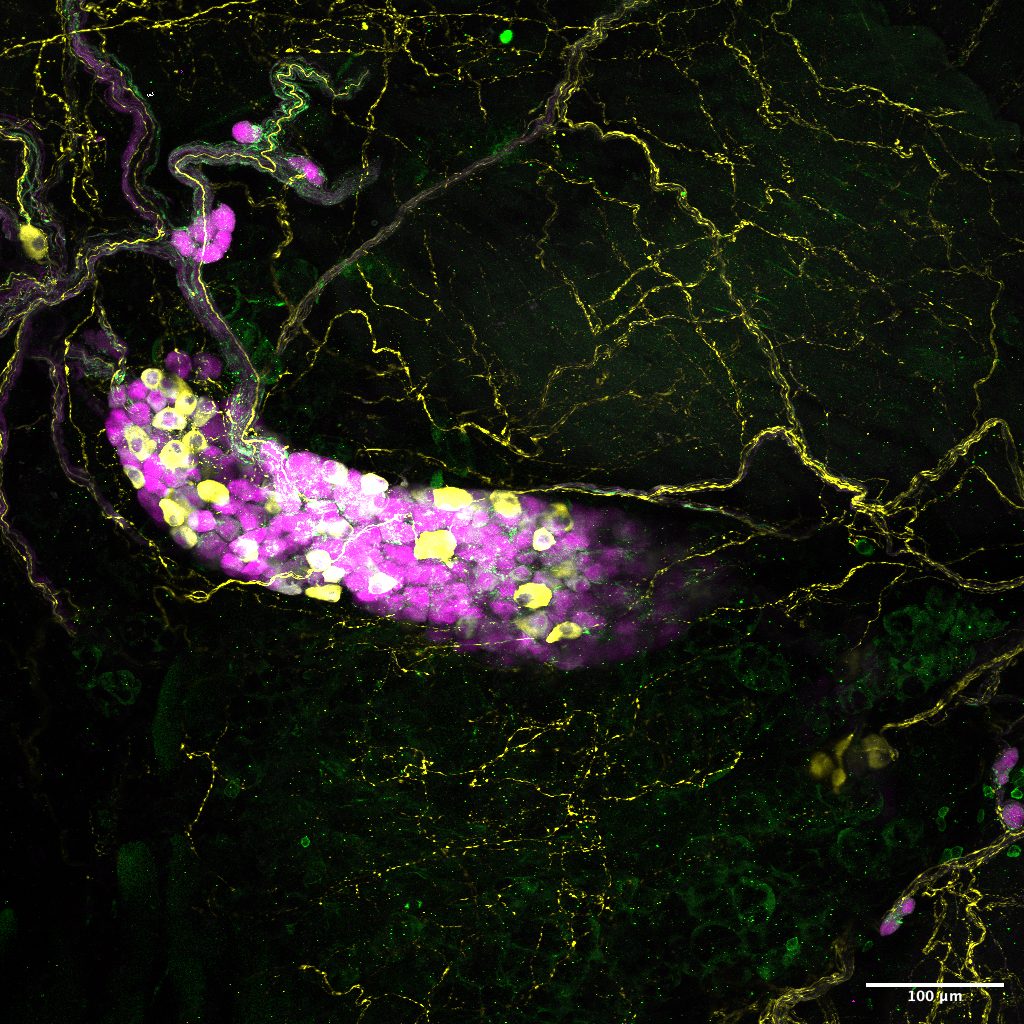
Different color stains label some of the diversity of neurons found in the mouse airway. Single-cell sequencing provides a means for distinguishing among the cell types and giving scientists genetic access to them. Image courtesy of Sara Prescott.
In her new work at MIT, she is not only following up on the functions of other vagal neurons, but also atlasing another novel population of neurons: the neurons intrinsic to the lungs and airways. No one has characterized them before.
“It’s shocking to me that we are still discovering cell types in the body,” Prescott said. “It’s really revolutionized a lot of cellular biology and molecular neuroscience.”
In a preprint posted in 2021, Heiman and Kellis produced one of the first atlases of the cell types of the human motor cortex. The survey of gene expression in 380,000 cells identified 46 distinct cell types. And in 2022 in Nature, they atlased the diversity of cells in the brain’s vasculature. They obtained more than 100 human postmortem samples, and 17 healthy brain tissue samples removed during surgery to treat epileptic seizures. They were therefore able to sequence more than 16,000 brain vasculature cells from people of different ages and genders. They discerned 11 distinct cell types, observed how gene expression changes based on where cells are in the vasculature (a property called “zonation”), and noted important differences in zonation between humans and mice that likely affect properties including what each species will allow to enter the brain through the blood-brain barrier.
Rather than a broad-scale atlas, Littleton’s recent studies focused on comparing gene expression between exactly two different cell types. They each connect to muscles to help control motion in a fly. His lab’s guiding question was why one type of neuron makes stronger connections, or synapses, than the other. Littleton’s lab extracted RNA from each type of cell in 100 flies, already knowing exactly which ones they were. His lab instead focused on determining how their gene expression differed and then investigating in the flies how those measured differences might matter to the cells’ synaptic properties. While many differences weren’t relevant, many were. In papers in Neuron and in Cell Reports this fall his lab not only reported the the important differences they’d discovered, but also showed that the cells edited their RNA extensively to produce even more fine-grained control over synaptic communication.
Mechanisms of disease
The power of single cell genomics to offer clues about how cells respond to disease has been the major focus of Tsai’s use of the technique in at least 10 studies over the last several years. In 2019 in Nature, she and Kellis published the first comprehensive analysis of differential gene expression in healthy and Alzheimer’s disease postmortem brains by sequencing 80,000 cells from 24 people who had Alzheimer’s disease and 24 otherwise similar people who did not. The research revealed that not only neurons but also microglia, astrocytes and oligodendrocytes showed important differences. In particular, oligodendrocytes have the responsibility of insulating the wiring that neurons grow to connect with each other. The study indicated that this insulation process, or “myelination,” was compromised. The data also showed sex differences in response to Alzheimer’s.
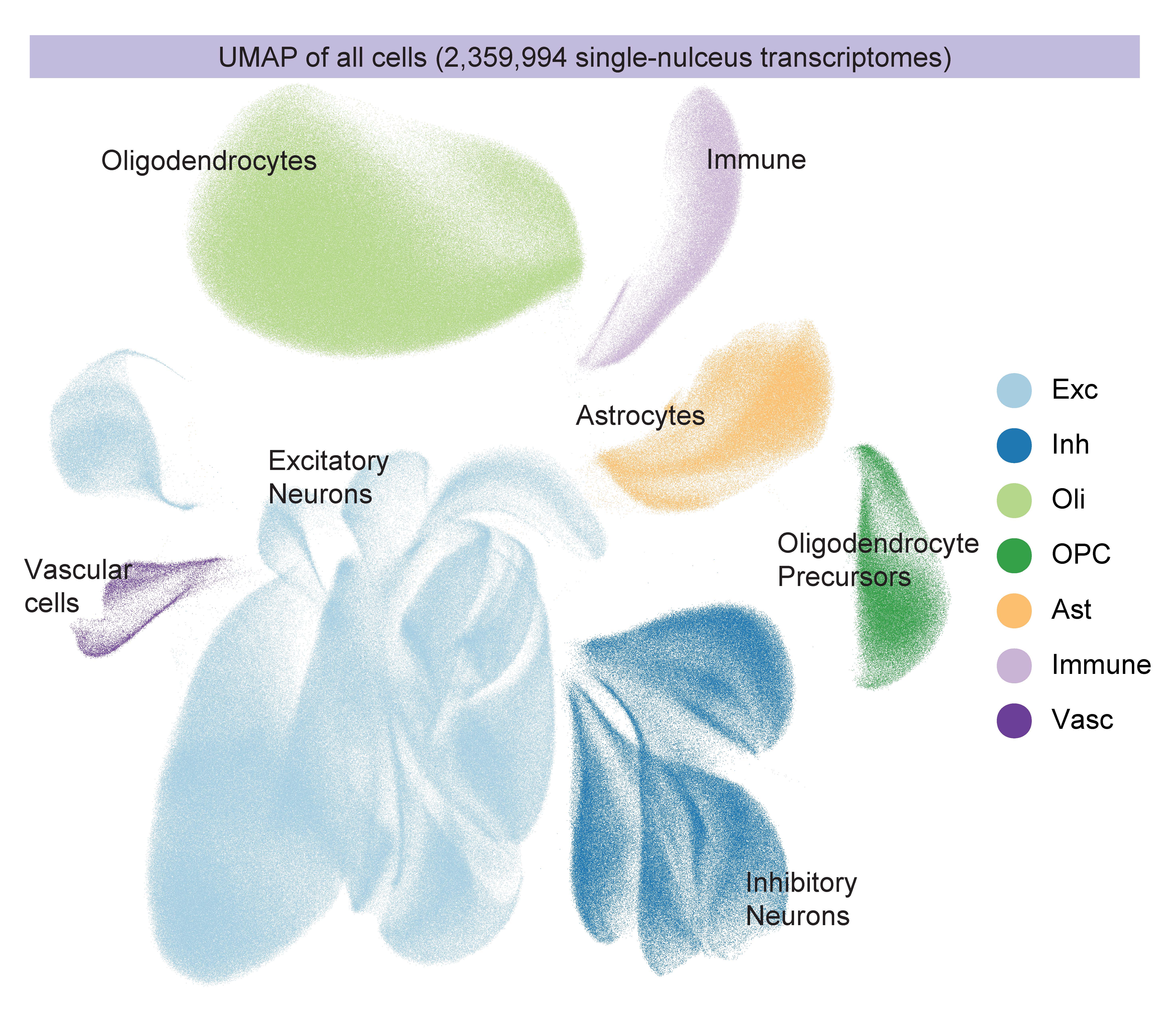
In 2023 the labs combined for a massive update. In four companion papers in Cell in September, the labs sequenced more than 2 million cells of 54 distinct types from 427 brain samples—an unprecedented effort. Through careful analysis and experimental follow-up, the labs revealed numerous insights including targets for potential therapies. Alzheimer’s disease afflicted cells struggled with mitochondrial function, lipid metabolism, synaptic signaling and maintenance of their genome. In one paper, however, they showed that when comparing people who remain cognitively resilient to those with cognitive impairment, a key difference was the relative abundance of specific types of inhibitory neurons.
A "UMAP" showing the categorical groupings of more than 2 million cells whose RNA was sequenced for Alzheimer's studies. Image by Hansruedi Mathys.
Another paper focused on how age-related lapses in DNA repair led to genome rearrangements and 3D folding defects. Yet another paper focused more deeply on microglia, showing that they assume 12 different states amid disease and that as more become inflammatory, it hinders both neural communication and the effectiveness of the blood brain barrier. But the study also pointed to transcription factors that, if altered, could reduce microglial inflammation. The fourth paper in the set combined scRNA-seq and scATAC-seq to delve into why some genes are expressed more than others and showed how the process goes awry in Alzheimer’s
In a 2023 study in Science Translational Medicine, Tsai’s lab incorporated scRNA-seq to pinpoint that an exact type of neuron in the brain’s mammillary body exhibits aberrant activity especially early in disease, leading to memory loss. And by comparing gene expression in cells with the biggest genetic risk factor for Alzheimer’s disease, the APOE4 gene variant, with those harboring the normal APOE3 version, the lab has identified how distinct cell types falter. For instance, in Nature in 2022, scRNA-seq helped Tsai’s lab show that APOE4 oligodendrocytes mishandle cholesterol, leading to myelination failures.
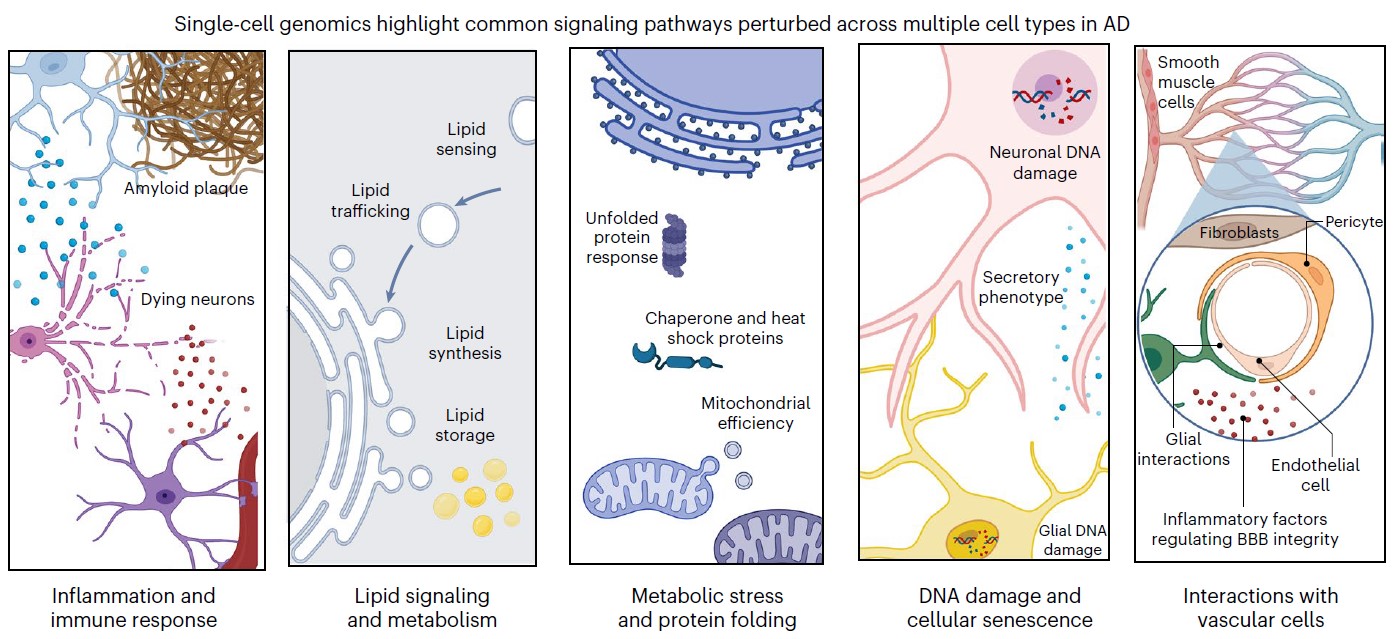
In the Nature Neuroscience review, Tsai and Murdock reported that single cell genomic studies from multiple labs worldwide have converged on five major pathways that become disturbed in major cell types in the Alzheimer’s disease (AD) brain.
Single-cell genomics by labs aroudn the world hve revealed five major common molecular pathways afected by Alzheimer's disease. Image by Mitch Murdock and Li-Huei Tsai.
“Single-cell profiling facilitates a nuanced portrait of the diverse cellular processes perturbed in the AD brain,” they wrote. “These varied molecular programs help explain the divergence between healthy aging and cognitive decline, and highlight cell-type-specific molecular programs involved in AD.”
Heiman’s scRNA-seq-aided studies have focused on other age-related neurodegenerative diseases. Her 2020 study in Neuron was one of the first to use the technique to characterize how cells respond to Huntington’s disease. The findings included that many aberrations in gene transcription may be related to a few factors that could be targeted with drugs. Another finding was that an especially vulnerable type of neuron mounts a misguided and potentially fatal innate immune response to unusual levels of mitochondrial RNA.
Signs of a similar problematic immune response also emerged in the 2022 study that atlased the vasculature. As part of that study, Heiman and Kellis compared gene expression in the vascular cells of people with and without Huntington’s disease. They found that an altered innate immune response and other factors may contribute to the blood-brain barrier becoming more permeable than it should be.
Similarly, the preprint featuring an atlas of the motor cortex features comparisons among the brains of people with ALS, Frototemporal Lobar Degeneration (FTLD), or neither of those motor disorders. Their findings included an interesting overlap: Betz cells (most vulnerable in ALS) and von Economo neurons (VENs; most affected in FTLD) were among the most affected in terms of gene expression, and turned out to have an almost identical basal gene expression profile,which might explain why the cells are especially vulnerable to the diseases.
In January 2023, Heiman teamed up with Kellis and Institute Professor Ann Graybiel in a study in Nature Communications that employed scRNA-seq to pinpoint how two distinct cell populations in a brain region called the striatum were affected differently by Huntington’s disease. The findings suggested that the disease’s damage to a population in the striatum’s matrix leads to motor impairments, while damage to the other population, located in structures called striosomes, may account for the mood disorders that manifest in early stages of the disease.
At the time, Heiman said: “This study addresses an important outstanding question in the field, how striosome-matrix striatal projection neuron identity is affected in Huntington’s disease. The use of single-cell RNA profiling has allowed us to address this question for the first time in a comprehensive manner.”
Across many studies in tissues as diverse as the lungs and regions deep in the brain, scRNA-seq has become a valuable tool.
Symposium speakers examine how understanding the brain could improve the functioning of democracy
June 2, 2026
Picower Events
Symposium speakers examine how understanding the brain could improve the functioning of democracy
Combining insights from the debates of American civics to the discoveries of neuroscience labs, experts honed in on ways that brain science research could inform efforts to improve political participation and dialogue in a polarized age
MIT-based team releases first AI foundation model for Alzheimer's prevention
April 26, 2026
Research Feature
MIT-based team releases first AI foundation model for Alzheimer's prevention
FINGERS-7B integrates lifestyle, clinical, genomic, and proteomic data from tens of thousands of at-risk individuals to discover multi-omic biomarkers for preclinical Alzheimer's
Alana Down Syndrome Center symposium highlights studies from brain to heart
April 14, 2026
Picower Events
Alana Down Syndrome Center symposium highlights studies from brain to heart
Seven researchers from MIT, Rutgers University and the University of São Paulo shared the research they are doing to help people with trisomy 21 throughout their lifespan.