
December 14, 2020
Their goal is to pinpoint how a genetic aberration undermines the function of brain cells and circuits. Learning how things go wrong puts within reach new ideas for preventing or reversing those problems. Indeed some strategies developed at the Picower Institute have advanced potential therapies all the way to clinical trials.
“Discovery of the gene is just the first step in a mechanism-based understanding and it can take decades,” says Mriganka Sur, Newton Professor of Neuroscience. Sur is a global leader in studying how mutations in the gene MECP2 hinders the development of neurons and neural circuits and the maturation of neural connections, called synapses, disrupting intellectual development in patients with Rett Syndrome, an autism-like disorder.
Because the brain is a uniquely complex system that constantly adapts and changes, diseases of the central nervous system can be especially tricky to understand mechanistically, especially when their pathology plays out over the course of decades, said Myriam Heiman, Associate Professor in the Picower Institute and the Broad Institute of MIT and Harvard.
In the early days of the Human Genome Project many people hoped that identifying genes would make finding treatments straightforward. Sometimes this is possible. In the brain-affecting metabolic disease PKU, for instance, identifying the genetically inherited loss of ability to regulate the biochemical phenylalanine led to a clear therapeutic strategy of restricting diet (though a cure remains undiscovered).
“But many diseases of the nervous system don’t follow that paradigm,” said Heiman, who this year has published two papers in Neuron finding novel mechanisms and potential therapeutic targets in Huntington’s disease, a fatal and incurable neurodegenerative disorder. Scientists have known for more than 25 years that just one type of mutation—an accumulation of repeating CAG trinucleotides in the Huntingtin gene—causes toxic aggregation of Huntingtin protein, but as Heiman’s new research shows, there is still a lot to learn about how that makes neurons vulnerable.
Genetic cause
Picower Professor Mark Bear has been waging a battle against Fragile X syndrome, the leading heritable cause of intellectual disability and autism, for nearly 20 years. Fragile X is caused by mutation—specifically CGG trinucleotide repeats—in the FMR1 gene that can eliminate production of the protein FMRP. As such it is a “simple” disease, Bear says.

“We actually know the cause,” he says. “We don’t have to guess about it. All that goes wrong in individuals with Fragile X syndrome is due to the absence of this FMRP protein.”
The work started back in 2002 with a paper in the Proceedings of the National Academy of Sciences (PNAS). In fundamental research on mechanisms of synaptic plasticity—the way neurons change their circuit connections in response to experience to enable development, learning and memory—Bear’s lab discovered that loss of FMRP exaggerates the consequences of activating a neurotransmitter receptor called metabotropic glutamate receptor 5, or mGluR5. Activating mGluR5 causes synapses to weaken, a normal process called long-term depression, but without the brake provided by FMRP, there is too much LTD.
With this mechanistic insight, Bear’s lab hatched a strategy of treating Fragile X by inhibiting mGluR5. They showed in Neuron in 2007 that it was enormously effective in mouse models and eventually brought a candidate drug to advanced clinical trials, but it fell short of meeting the clinical goals set by regulators.
Undaunted, earlier this year Bear’s team published a new study in Science Translational Medicine (STM) demonstrating a promising updated approach. Study data indicated that one potential problem was that patients could build up a tolerance to the mGluR5 medicine. So Bear’s lab pursued another target in the mGluR5 pathway: the enzyme GSK3-alpha. Working with scientists at the Broad Institute, they tested a new drug to inhibit the enzyme, which is overactive in Fragile X mouse models, and found that doing so successfully reversed several symptoms. The drug showed no concerning side effects in the mice and the rodents did not build up tolerance.
“We don’t know whether the mGluR5 trials failed because of treatment resistance, but it’s a viable hypothesis,” Bear says. “What we do know is with the GSK3 alpha inhibitor, we do not see that in mice.”
Sur’s quest to treat Rett syndrome has some parallels with Bear’s experience. He, too, happened upon a promising treatment strategy by performing fundamental research on synaptic plasticity (which is a core research theme at The Picower Institute).

MECP2’s protein regulates the expression of hundreds of other genes. In Nature Neuroscience in 2006, Sur’s lab discovered evidence that a protein called IGF1 is a crucial component in how synapses in the visual cortex in newborn mice develop in response to visual activity. IGF1 is regulated by MECP2 and is indeed notably reduced in mouse models of Rett Syndrome. Sur’s lab has shown that the lack of IGF1, in turn, reduces levels of proteins such PI3K/Akt, and that undermines still other proteins needed for synapses to strengthen and mature properly. In a paper in 2014 in PNAS the team showed that treating mice by dosing them with IGF1 corrects functional, structural, and molecular mechanisms downstream of MECP2 and improves mouse behavior. Since then, the idea has been tested in Rett syndrome patients in clinical trials. One trial did not show sufficient efficacy, but another phase III study is still pending.
Sur’s lab has not stopped working on the problem. Several lab members continue to study how the gene regulation and expression cascade that depends on MECP2 affects neural development, not only in mice but also in complex 3D models of human brain tissue called cerebral organoids. And in STM last year, Sur collaborated with MIT biologist Rudolf Jaenisch of the Whitehead Institute to show that increasing expression of another key protein in the cascade, KCC2, also provided therapeutic benefits in mouse models, offering a fresh lead.
“Nothing has worked in patients yet, but you know we have to keep trying,” Sur said. “It’s a very hard problem. It goes to the heart of how the brain is made, which is a very complex process.”
So, as it turns out, is the process by which the brain is unmade in neurodegenerative disorders such as Huntington’s disease.

In her work to understand the mechanisms by which the huntingtin mutation leads to the deaths of specific neurons in a brain region called the striatum, Heiman has been using new, unbiased techniques to study the brains of Huntington’s disease patients and also those of mouse models of the disease. Unbiased means she isn’t confining her studies to prior hypotheses because doing so would be to assume that the field already knows everything that’s going wrong. But in diseases of the aging brain, it is very hard to fully understand the drivers of disease pathology, Heiman said. Decades of damage and compensation can obscure underlying mechanisms. She likens it to examining an unprotected crime scene.
“There have been many mechanisms put forward in the past, but they haven’t been successful targets in clinical trials,” Heiman said.
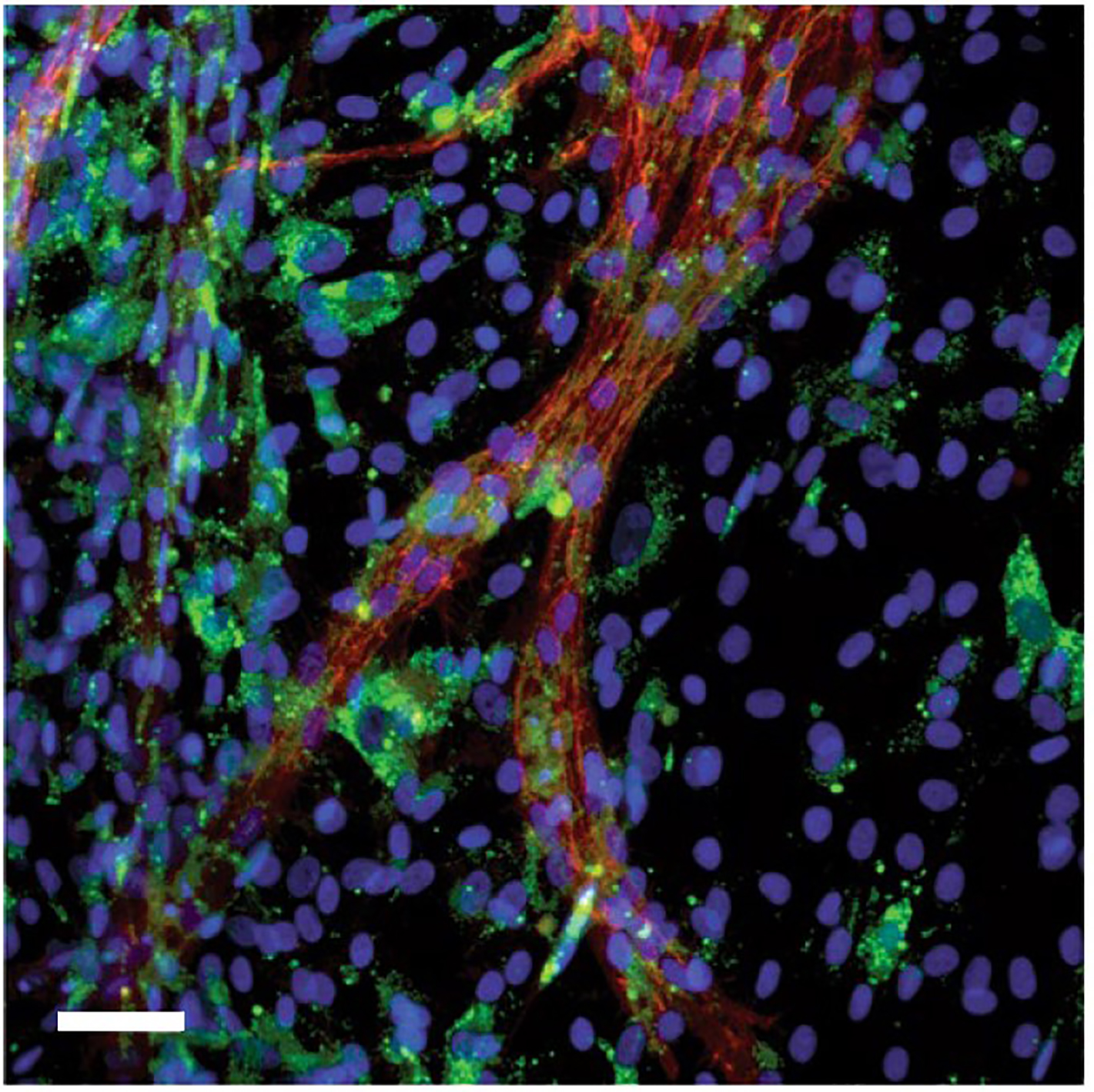
In January she published a study in which her lab knocked out all 22,000 genes found in the mouse striatum in a way in which any given cell had only about one gene knocked out. No one had ever accomplished this feat before in a mammal’s brain, but by doing so Heiman’s team was able to learn which genes are necessary for neurons to survive. When her team did the screen in Huntington’s disease model mice, they identified a particular gene family, called Nme, that had never been associated with Huntington’s before, but which was clearly necessary for neurons to survive the disease. The gene, as it turns out, may help cells dispose of protein aggregates (such as mutant Huntingtin). Heiman’s team showed that when Nme1 is overexpressed in the mouse models of Huntington’s, disease symptoms appear to improve.
Then in July, Heiman’s lab produced another study based on unbiased profiling of gene expression in striatal neurons known to be vulnerable in the disease. In this case, the researchers looked at gene transcription and translation (as measured by levels of RNAs) in different types of cells in both human and mouse brains. Across both species, and across different stages of the disease, they found that RNA from mitochondria were misplaced in striatal cells, called spiny projection neurons, that are particularly vulnerable to dying. They found evidence that the brain cells were perishing amid an immune response to the errant mitochondrial RNA, which looked to the cells like a viral invader. This insight offers another new mechanism of Huntington’s pathobiology that could be targeted in the future.
Genetic risk factors
On the other side of the Picower Institute’s fourth floor, Picower Professor and Institute director Li-Huei Tsai is hunting for therapies for a different neurodegenerative disease: Alzheimer’s. Her lab takes many approaches, but among is understanding the mechanistic failures brought about by the disease’s single biggest genetic risk factor, the APOE4 variant of the APOE gene.

As with autism spectrum disorders, the vast majority of Alzheimer’s cases cannot be traced to a clear inherited genetic cause (Fragile X and Rett syndromes are exceptions). Instead, most Alzheimer’s is associated with variations in many genes, which are not causal but appear to convey extra risk. For that information to be useful, scientists need to what effect the variations have.
Over the last few years, Tsai’s lab has revealed several things that go wrong in brain cells carrying APOE4 vs. the more common APOE3 version. In 2018, they used stem cells derived from patients’ skin cells to create cultures of key brain cell types: neurons, microglia and astrocytes. In Neuron the team showed that APOE4 neurons secreted more amyloid protein, a hallmark of Alzheimer’s pathology, than APOE3 neurons. Meanwhile, APOE4 astrocytes and microgllia showed less ability to remove amyloid.
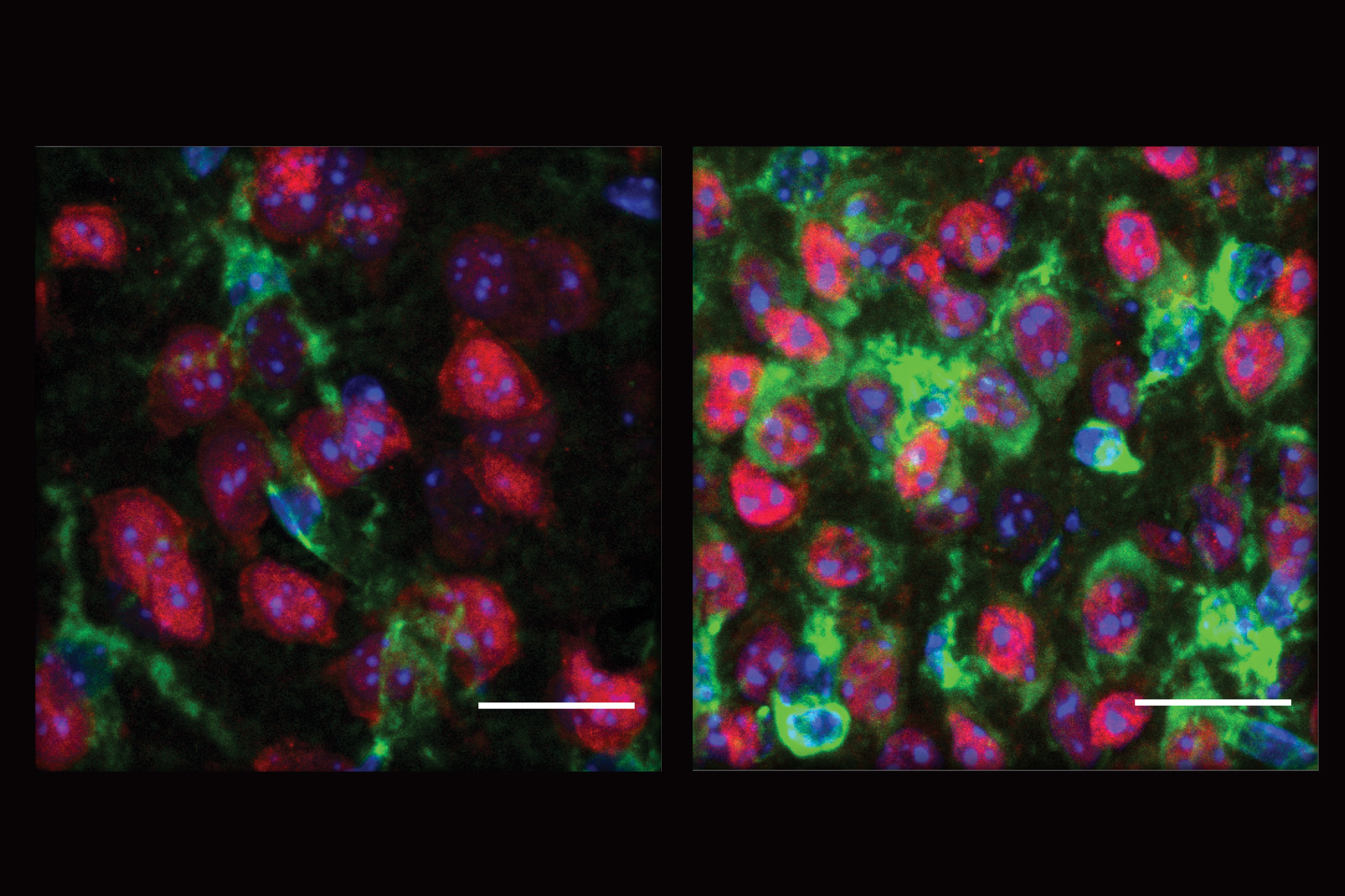
In a study earlier this year in Nature Medicine, Tsai’s lab used similar techniques to engineer APOE3 and APOE4 versions of the blood-brain barrier (BBB), the security fence that keeps unwanted pathogens or chemicals from entering the brain. The BBB is almost always disrupted in Alzheimer’s by a strongly correlated condition called Cerebral Amyloid Angiopathy. Tsai’s team was able to show that APOE4 made the BBB more vulnerable particularly because of disrupted functioning among specific vascular cells called pericytes. The scientists went on to identify the specific mechanistic pathway that becomes errant and that it can be corrected with specific medications.
A few months later, Tsai’s lab published yet another set of findings about APOE4’s ill-effect in the brain. They showed that astrocytes with APOE4 appeared to struggle with the crucial process of endocytosis, a major means by which the cells bring material into their bodies, which in turn can undermine how they support healthy brain function. This study took a hopeful turn, however, by showing that overexpression of another Alzheimer’s associated gene, PICALM, corrected the defect.
Not only was the study an important advance in understanding how APOE4 may undermine brain function, and maybe how to treat it, but also it highlighted the value of an emerging strategy in studying Alzheimer’s disease and assessing it in patients: polygenic risk. Scientists are beginning to realize that they need to know how genetic variants may combine and accumulate to affect disease risk for patients, Tsai said.
“It’s a concept people have talked about for a long time, but I think now people are finally beginning to think seriously about how to work on it,” Tsai said. “Someone’s risk for developing Alzheimer’s is probably in large part based on their genetics—how many risk alleles they carry.”
The same is likely true in bipolar disorder, said Elly Nedivi, William R. and Linda R. Young Professor of Neuroscience in the Picower Institute. In 2019 in Molecular Psychiatry, she showed how single and combinations of variants in the gene SYNE1 appear to undermine the ability to regulate synaptic strength in bipolar disorder. However, none of these variants which affect expression or function of CPG2, a key protein encoded by SYNE1, are sufficient to outright cause bipolar disorder on their own. Instead they likely increase a susceptibility that is influenced by many factors – much like family history contributes to the chance of developing heart disease, but is not deterministic on its own.

“It’s a risk—its something that’s against you, but the question is what else do you have going for you?,” Nedivi said. “The constellation of SYNE1 variants in combination with the rest of your genetic makeup, as well as other factors in your life will determine together whether you will develop the disease.”
Nedivi’s lab discovered CPG2 while screening neurons for proteins whose expression depends on synaptic activity. Their 2004 study in Neuron revealed that CPG2 is expressed in an area right next to excitatory synapses that helps to traffic receptors for the neurotransmitter glutamate to the synapse. As such, it is important for helping synapses adapt to activity levels by adding or removing these receptors.
In the 2019 study, Nedivi’s research team identified several SYNE1 variants in people with biopolar disorder and tested the effect of these variants on CPG2 level and function in rat neurons. They found that the different genetic aberrations undermined CPG2 in various ways— by reducing its expression, or attenuating its ability to regulate glutamate receptor trafficking, particularly in response to changes in synaptic transmission, when it’s most needed.. The study was the first to connect the finding of genetic factors in bipolar to a specific physiological mechanism in the brain.
Nedivi’s lab is following up on the findings. This fall she hired a new postdoctoral researcher to continue this work, in particular to test whether the detrimental affects of bipolar patient mutations in CPG2 can be counteracted pharmaceutically.
“I love this project,” Nedivi said. “I think it has enormous potential for both therapeutics, as well as diagnostics.”
After all, as in the other ongoing efforts of Picower Institute researchers, it represents the rewarding possibility—even if it’s a steep climb—of going from identifying genes, to developing neuroscientific understanding, to treating devastating diseases.



