
For decades, biologists have performed genetic screens in which they systematically knock out individual genes in animal models and then observe the effects on cell survival. But this approach has never been feasible in the mammalian brain because of the technical difficulty of delivering the needed genetic materials to the brain.
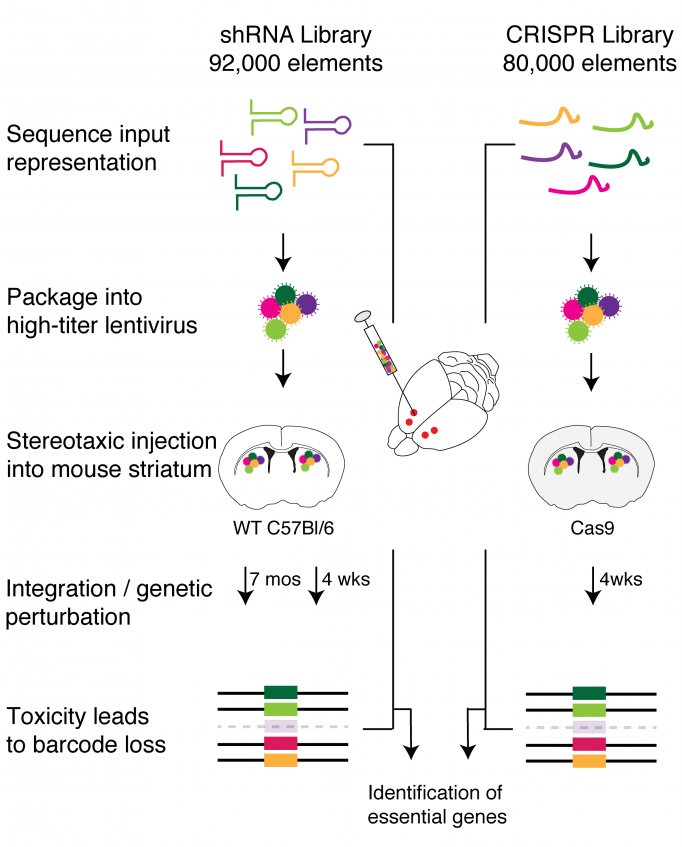
Recently researchers at the Broad Institute of MIT and Harvard have developed libraries of genetic material (elements using either CRISPR or short hairpin RNA) that can be used to turn off the expression of each of the 22,000 genes found in the mouse genome. The libraries are delivered by viruses, each of which carry one element to a cell to target a single gene.
For a study published in Neuron in January 2020, Myriam Heiman’s team at The Picower Institute and the Broad Institute came up with a way to highly concentrate these viruses and to inject them directly into the striatum of the mouse brain, a brain region implicated in Huntington’s disease, Parkinson’s disease, and various neuropsychiatric disorders. Using this approach, they were able to deliver the screening libraries efficiently into the cells of the striatum. After incubating in the brain, the researchers sequenced all of the genomic DNA in the targeted striatal neurons. The idea was that if particular genes are necessary for neurons’ survival, any cell with those genes knocked out will die. Then, those shRNAs or CRISPR elements will be found at lower rates in the total population of cells. Their study thereby achieved the first unbiased genome-wide knockdown screening in the mammalian central nervous system.
Heiman’s team applied this screen in two different mouse models of Huntington’s disease (in which the huntingtin protein forms harmful clumps) and compared the results from the screen of the Huntington’s model mice to normal mice. When the shRNA or CRISPR elements were found less frequently in the Huntington’s model mice, that indicated that those elements targeted genes that were helping to make cells more resistant to huntingtin’s the toxic effects. Using this method the team found a novel target: The Nme1 gene. Subsequent testing in the mice has suggested that increasing Nme1 activity could have therapeutic value for Huntington’s patients.
In 2021, Heiman earned an award from The Mathers Foundation to apply the technique to the study of Parkinson's disease.