Rett syndrome is a severe, autism-like neurodevelopmental disorder in girls characterized by intellectual disability and other neurological and non-neurological symptoms. It is caused by mutations in the MeCP2 gene on the X chromosome. In fundamental research on the proteins crucial for allowing neural connections, or synapses, to change with experience (a property called “plasticity”), the lab of Mriganka Sur, Newton Professor of Neuroscience, found that the protein IGF1, which is regulated by MeCP2, could remedy many of the symptoms of Rett syndrome in mouse models if artificially restored to healthy levels. A pharmaceutical company has acted on the lab’s results, testing a therapy based on the idea in Phase III clinical trials that concluded with positive results last year.
The lab discovered the role of IGF1 in neural plasticity in 2006 when they showed that it critically affected the ability of the visual cortex to shift connections away from supporting a closed eye in favor of one left open (a dramatic phenomenon called ocular dominance plasticity).
In 2009 they revealed that IGF1 was also consequential to Rett syndrome. IGF1 becomes misregulated by the loss of MeCP2. In the study the team showed that dosing Rett model mice with extra IGF1 peptide fragment improved many disease symptoms and also enhanced neural plasticity in the mice, promoting improved brain function. In 2014 they complemented those results using doses of recombinant human IGF1. Encouraging results of the first human clinical trial of IGF1 for Rett syndrome appeared that same year with Sur as a co-author.
In 2016 the lab produced another study finding that a critical problem for neural function in MeCP2 deficient mice was that neurons responsible for computations received too little excitatory and inhibitory drive from others, a circumstance they were able to repair with doses of IGF1.
In addition to identifying IGF1, the lab has also produced other key insights into Rett syndrome. In 2017 using advanced human tissue cultures, the lab found that in the absence of MeCP2, certain microRNAs critical to proper brain development become misregulated. They found that overexpression of the microRNAs prevented new neurons from being born and that inhibiting the microRNAs enabled neural birth.
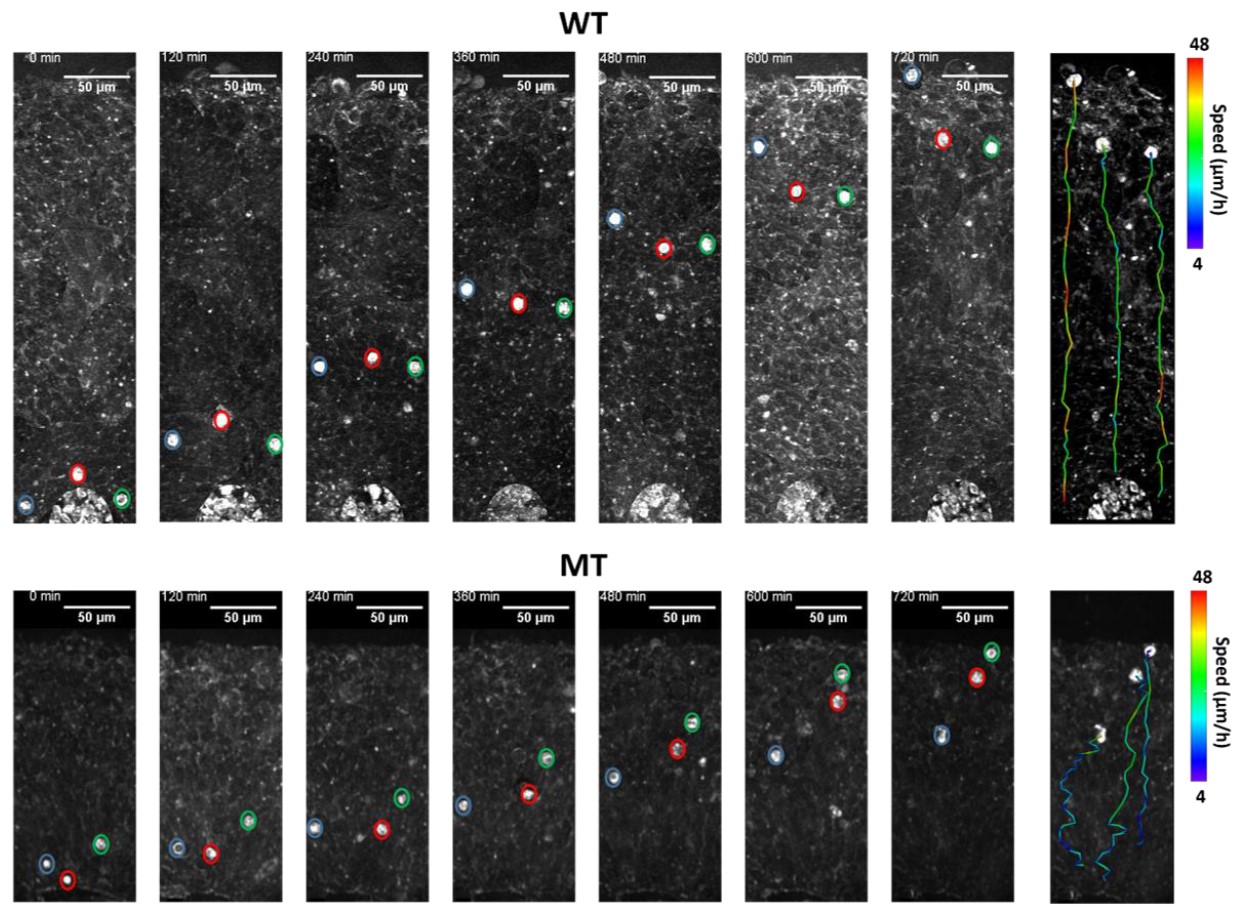
Most recently, they used a combination of advanced imaging methods and 3-dimensional tissue culture techniques used to grow cerebral cortex-like structures (‘organoids’) to show that a core pathology of Rett syndrome arose from altered migration of neurons as they settled into the cerebral cortex. Thus, Rett syndrome is characterized by a host of factors during development that lead to altered brain function.